Das könnte Ihnen auch gefallen
- Amplicon Melting Analysis With Labeled Primers - A Closed-Tube Method For Differentiating Homozygotes and HeterozygotesDokument12 SeitenAmplicon Melting Analysis With Labeled Primers - A Closed-Tube Method For Differentiating Homozygotes and HeterozygotesShavana RajkumarNoch keine Bewertungen
- Molecular Pathology Techniques: Clinics in Laboratory Medicine December 2013Dokument21 SeitenMolecular Pathology Techniques: Clinics in Laboratory Medicine December 2013Nguyễn HuyềnNoch keine Bewertungen
- Comparative Diagnostic Methods For Canine EhrlichiosisDokument9 SeitenComparative Diagnostic Methods For Canine EhrlichiosisNeny ChalondokterNoch keine Bewertungen
- Quantifying RNA With QRT-PCR: 2-3 HoursDokument12 SeitenQuantifying RNA With QRT-PCR: 2-3 Hourszigurat00Noch keine Bewertungen
- Rapid Cytokine SNP Analysis Using LightCycler TechnologyDokument2 SeitenRapid Cytokine SNP Analysis Using LightCycler TechnologyGanesh JoNoch keine Bewertungen
- Modern Molecular MethodsDokument15 SeitenModern Molecular MethodsYeo Wee KiatNoch keine Bewertungen
- Barry Schweitzer Et Al - Immunoassays With Rolling Circle DNA Amplification: A Versatile Platform For Ultrasensitive Antigen DetectionDokument7 SeitenBarry Schweitzer Et Al - Immunoassays With Rolling Circle DNA Amplification: A Versatile Platform For Ultrasensitive Antigen DetectionGmso3Noch keine Bewertungen
- Desviat 2006 CcaDokument4 SeitenDesviat 2006 Ccarahmawati aliwarmanNoch keine Bewertungen
- Search Database Search TermDokument31 SeitenSearch Database Search Termorode franklynNoch keine Bewertungen
- Parade LaDokument6 SeitenParade Laptrocha21Noch keine Bewertungen
- DNA-based meat speciation techniquesDokument4 SeitenDNA-based meat speciation techniquesAhmed J AlhindaweNoch keine Bewertungen
- JCM 40 10 3814-3817 2002Dokument4 SeitenJCM 40 10 3814-3817 2002ERIE YUWITA SARINoch keine Bewertungen
- LOD, LOQ, Dynamic Range of Real Time PCRDokument12 SeitenLOD, LOQ, Dynamic Range of Real Time PCRNguyen Phuong LoanNoch keine Bewertungen
- Koo Ken 2014Dokument7 SeitenKoo Ken 2014Perla Andrea Urriola AraosNoch keine Bewertungen
- Allele Frequencies of Six MiniSTR Loci in The Population of Northern PortugalDokument3 SeitenAllele Frequencies of Six MiniSTR Loci in The Population of Northern PortugalspanishvcuNoch keine Bewertungen
- Pharmaceutical Applications of GCDokument20 SeitenPharmaceutical Applications of GCAlinaDianaNoch keine Bewertungen
- Basic and Advanced Laboratório Techniques in Histopathology and CytologyDokument11 SeitenBasic and Advanced Laboratório Techniques in Histopathology and CytologyCláudia TorresNoch keine Bewertungen
- Chrom PaparDokument8 SeitenChrom Paparvinodk.ormindiaNoch keine Bewertungen
- ToxigcmsDokument37 SeitenToxigcmsTadesse TakeleNoch keine Bewertungen
- AP1 AP2 PCR Method For AHPND Detection - Flegel 2013Dokument6 SeitenAP1 AP2 PCR Method For AHPND Detection - Flegel 2013Alden ReynaNoch keine Bewertungen
- Applications of Eco-Real Time PCRDokument4 SeitenApplications of Eco-Real Time PCRShubhendu SealNoch keine Bewertungen
- Jurnal RFLP PDFDokument8 SeitenJurnal RFLP PDFSilvia ZahiraNoch keine Bewertungen
- Quantitative Real-Time PCR: A Powerful Ally in Cancer ResearchDokument7 SeitenQuantitative Real-Time PCR: A Powerful Ally in Cancer ResearchFaridRinaddinNoch keine Bewertungen
- Determination of Haplotypes From Single DNA Molecules: A Method For SingleMolecule BarcodinDokument9 SeitenDetermination of Haplotypes From Single DNA Molecules: A Method For SingleMolecule BarcodinMatt GordonNoch keine Bewertungen
- Tandem Dyes For Flow Cytometry, Quality Concerns, Beckman CoDokument2 SeitenTandem Dyes For Flow Cytometry, Quality Concerns, Beckman CocandiddreamsNoch keine Bewertungen
- Drugs and Poisons in Humans MethodeDokument11 SeitenDrugs and Poisons in Humans MethodeFaeyza Abimanyu SNoch keine Bewertungen
- Nar00093 0083Dokument7 SeitenNar00093 0083Marfalab LaboratorioNoch keine Bewertungen
- 91 120 1 PBDokument6 Seiten91 120 1 PBSudarmono Ahmad TahirNoch keine Bewertungen
- Anal BiochemDokument7 SeitenAnal BiochemRonna Selomenio AlarconNoch keine Bewertungen
- Detection of bacteria in clinical samples using 16S rRNA gene sequencingDokument6 SeitenDetection of bacteria in clinical samples using 16S rRNA gene sequencingrehanaNoch keine Bewertungen
- Race PCR DissertationDokument8 SeitenRace PCR DissertationWriteMyPapersDiscountCodeUK100% (1)
- Study of XPD Gene Polymorphism in HCV Positive HCC Egyptian PatientsDokument4 SeitenStudy of XPD Gene Polymorphism in HCV Positive HCC Egyptian PatientsdrosabryNoch keine Bewertungen
- Uniparental Disomy (UPD) Analysis of Chromosome 15: Applied Biosystems 3500xL Genetic AnalyzerDokument6 SeitenUniparental Disomy (UPD) Analysis of Chromosome 15: Applied Biosystems 3500xL Genetic AnalyzerPedro Guerrero PérezNoch keine Bewertungen
- Isolation and Purification of Polyclonal Igg Antibodies From Bovine Serum by High Performance Liquid ChromatographyDokument7 SeitenIsolation and Purification of Polyclonal Igg Antibodies From Bovine Serum by High Performance Liquid ChromatographyMoody ModmodNoch keine Bewertungen
- Approximately 8,000 New Molecular Markers Generated for Chlamydomonas reinhardtiiDokument4 SeitenApproximately 8,000 New Molecular Markers Generated for Chlamydomonas reinhardtiiAlvisha PatwardhanNoch keine Bewertungen
- Mathematical Considerations of Competitive Reaction Polymerase ChainDokument11 SeitenMathematical Considerations of Competitive Reaction Polymerase ChainNatália RochaNoch keine Bewertungen
- 5.1.4 Gas Chromatography: 13 Changing Trends in The Methodologies of Extraction and Analysis of - . - 397Dokument31 Seiten5.1.4 Gas Chromatography: 13 Changing Trends in The Methodologies of Extraction and Analysis of - . - 397Raque PcNoch keine Bewertungen
- Graphite Furnace Atomic Absorption Spectroscopic Measurement of Blood Lead in Matrix-Matched StandardsDokument5 SeitenGraphite Furnace Atomic Absorption Spectroscopic Measurement of Blood Lead in Matrix-Matched StandardsSaskhia GomezNoch keine Bewertungen
- Molecular Methods in Diagnosis of Infectious DiseasesDokument68 SeitenMolecular Methods in Diagnosis of Infectious DiseasesPeachy Pie100% (1)
- Olerup QTYPE HLA Typing Kits IFU v9-3Dokument19 SeitenOlerup QTYPE HLA Typing Kits IFU v9-3huripNoch keine Bewertungen
- Forensic Science International 132 (2003) 76-81 PDFDokument6 SeitenForensic Science International 132 (2003) 76-81 PDFJe RivasNoch keine Bewertungen
- Characterization of The New Serum Protein Reference Material Erm Da470k Ifcc Value Assignment by ImmunoassayDokument9 SeitenCharacterization of The New Serum Protein Reference Material Erm Da470k Ifcc Value Assignment by Immunoassaypatricia.mprpNoch keine Bewertungen
- Molecular Basis of A Progressive Juvenile-Onset Hereditary CataractDokument6 SeitenMolecular Basis of A Progressive Juvenile-Onset Hereditary Cataractnugroho2212Noch keine Bewertungen
- Analytical Biochemistry: Claudia Lefimil, Carla Lozano, Irene Morales-Bozo, Anita Plaza, Cristian Maturana, Blanca UrzúaDokument3 SeitenAnalytical Biochemistry: Claudia Lefimil, Carla Lozano, Irene Morales-Bozo, Anita Plaza, Cristian Maturana, Blanca UrzúaWill BustNoch keine Bewertungen
- Mass Spectrometry of Nucleic AcidsDokument2 SeitenMass Spectrometry of Nucleic AcidsnbkondaNoch keine Bewertungen
- Meth Impurity ProfilingDokument16 SeitenMeth Impurity Profilingconker4Noch keine Bewertungen
- Presentation HPLCDokument81 SeitenPresentation HPLCSangram KendreNoch keine Bewertungen
- Submitted by Palash Ghosh M.SC Biotechnology Amity University, Haryana 2017Dokument29 SeitenSubmitted by Palash Ghosh M.SC Biotechnology Amity University, Haryana 2017Palash GhoshNoch keine Bewertungen
- Mycobacterium Tuberculosis Clinical Isolates UsingDokument8 SeitenMycobacterium Tuberculosis Clinical Isolates UsingMonica Sari SuryadiNoch keine Bewertungen
- A Fluorescent SNP Detection Based On The Graphene Platform: Preparation and Characterization of GODokument4 SeitenA Fluorescent SNP Detection Based On The Graphene Platform: Preparation and Characterization of GOFamiloni LayoNoch keine Bewertungen
- Polymerase Chain Reaction PCRDokument4 SeitenPolymerase Chain Reaction PCRsister girlfriendNoch keine Bewertungen
- Design, Expression and Characterization of A Highly Stable Tetratricopeptide-Based Protein Scaffold For Phage Display ApplicationDokument6 SeitenDesign, Expression and Characterization of A Highly Stable Tetratricopeptide-Based Protein Scaffold For Phage Display ApplicationsuryasanNoch keine Bewertungen
- Group 4: DIGITAL PCRDokument14 SeitenGroup 4: DIGITAL PCRMaria amor MacapallagNoch keine Bewertungen
- Modern Analytical TechniquesDokument5 SeitenModern Analytical TechniquesRia SinghNoch keine Bewertungen
- Comparison of SLCO1B1 Sequence Variability Among GermanDokument10 SeitenComparison of SLCO1B1 Sequence Variability Among GermanRicardo Perea JacoboNoch keine Bewertungen
- Stojkovic 2012Dokument5 SeitenStojkovic 2012Marko StojkovicNoch keine Bewertungen
- B1 - Artikel International Journal of Engineering and Technology Ema Komalasari - Penulis 1Dokument7 SeitenB1 - Artikel International Journal of Engineering and Technology Ema Komalasari - Penulis 1Wulan DariNoch keine Bewertungen
- Deshmukh 2000Dokument24 SeitenDeshmukh 2000Tecno QB7Noch keine Bewertungen
- Analytical Methods for Major and Modified Nucleosides - HPLC, GC, MS, NMR, UV and FT-IRVon EverandAnalytical Methods for Major and Modified Nucleosides - HPLC, GC, MS, NMR, UV and FT-IRNoch keine Bewertungen
- ISO Standards and Bias Pierce DrorDokument10 SeitenISO Standards and Bias Pierce DrorchiralicNoch keine Bewertungen
- Microsart @media: Advanced System For Touch-Free Membrane TransferDokument4 SeitenMicrosart @media: Advanced System For Touch-Free Membrane TransferVijay Kumar NandagiriNoch keine Bewertungen
- Data Microsart Ejet SM 2008 A DataDokument2 SeitenData Microsart Ejet SM 2008 A DatachiralicNoch keine Bewertungen
- Head Space Gas Chromatography Analysis of Residual Solvents in Temozolomide by Using Zb-624 ColumnDokument9 SeitenHead Space Gas Chromatography Analysis of Residual Solvents in Temozolomide by Using Zb-624 ColumnchiralicNoch keine Bewertungen
- KAM LKF and PKF: Karl Fischer Moisture AnalyzerDokument22 SeitenKAM LKF and PKF: Karl Fischer Moisture AnalyzerchiralicNoch keine Bewertungen
- Drugs 1Dokument14 SeitenDrugs 1Theesha SophieNoch keine Bewertungen
- Fungi: Candida Auris: A Review of Recommendations For Detection and Control in Healthcare SettingsDokument11 SeitenFungi: Candida Auris: A Review of Recommendations For Detection and Control in Healthcare SettingschiralicNoch keine Bewertungen
- Microsart Manifolds: The Filtration Stand That Adapts To Your NeedDokument8 SeitenMicrosart Manifolds: The Filtration Stand That Adapts To Your NeedchiralicNoch keine Bewertungen
- Plastics Identification Flow ChartDokument1 SeitePlastics Identification Flow ChartchiralicNoch keine Bewertungen
- A Review On Recent Developments in The Determination of Residual Solvents by Gas ChromatographyDokument9 SeitenA Review On Recent Developments in The Determination of Residual Solvents by Gas ChromatographychiralicNoch keine Bewertungen
- The World Clock - WorldwideDokument1 SeiteThe World Clock - WorldwidechiralicNoch keine Bewertungen
- On-Site Fuel and Lubricant Quality Control by Portable FTIR AnalysersDokument2 SeitenOn-Site Fuel and Lubricant Quality Control by Portable FTIR AnalyserschiralicNoch keine Bewertungen
- Application: Organic Volatile Impurities in Pharmaceutical Products: Selectivity of Capillary GC ColumnsDokument2 SeitenApplication: Organic Volatile Impurities in Pharmaceutical Products: Selectivity of Capillary GC ColumnschiralicNoch keine Bewertungen
- Candida auris Review: Emerging Multidrug PathogenDokument7 SeitenCandida auris Review: Emerging Multidrug PathogenchiralicNoch keine Bewertungen
- Simultaneous Estimation of Residual Solvents (Ethanol, Acetone, Dichloromethane and Ethyl Acetate) in Dosage Form by GC-HS-FIDDokument7 SeitenSimultaneous Estimation of Residual Solvents (Ethanol, Acetone, Dichloromethane and Ethyl Acetate) in Dosage Form by GC-HS-FIDchiralicNoch keine Bewertungen
- Attune NXT Flow Cytometer: Efficient. Flexible. TransformativeDokument36 SeitenAttune NXT Flow Cytometer: Efficient. Flexible. TransformativechiralicNoch keine Bewertungen
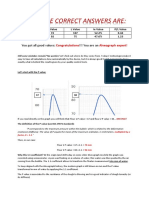
- And The Correct Answers Are:: You Got All Good Values: !!! You Are AnDokument3 SeitenAnd The Correct Answers Are:: You Got All Good Values: !!! You Are AnchiralicNoch keine Bewertungen
- Grain and Processed Grain Solutions: The Complete Product RangeDokument8 SeitenGrain and Processed Grain Solutions: The Complete Product RangechiralicNoch keine Bewertungen
- Baths - Chillers - Resource GuideDokument8 SeitenBaths - Chillers - Resource GuidechiralicNoch keine Bewertungen
- Application Note - HiQ Hydrogen As An Alternative To Helium For GC - tcm899-90120Dokument16 SeitenApplication Note - HiQ Hydrogen As An Alternative To Helium For GC - tcm899-90120chiralicNoch keine Bewertungen
- AN362Dokument2 SeitenAN362chiralicNoch keine Bewertungen
- Electrodes For Titration: Which Electrode For Which Application?Dokument2 SeitenElectrodes For Titration: Which Electrode For Which Application?chiralicNoch keine Bewertungen
- Electrodes For Titration: Which Electrode For Which Application?Dokument2 SeitenElectrodes For Titration: Which Electrode For Which Application?chiralicNoch keine Bewertungen
- Quality Control Solutions For The Feed IndustryDokument4 SeitenQuality Control Solutions For The Feed IndustrychiralicNoch keine Bewertungen
- Bax System q7 Mpb-2001Dokument8 SeitenBax System q7 Mpb-2001Tlw QuirogaNoch keine Bewertungen
- Application Compare Sipper Cuvettes Cary-3500 Uv-Vis 5994-1951en Us AgilentDokument6 SeitenApplication Compare Sipper Cuvettes Cary-3500 Uv-Vis 5994-1951en Us AgilentchiralicNoch keine Bewertungen
- CO Incubator: Resource GuideDokument8 SeitenCO Incubator: Resource GuidechiralicNoch keine Bewertungen
- 425-06 - Duo Sas 360 - UkDokument1 Seite425-06 - Duo Sas 360 - UkchiralicNoch keine Bewertungen
- HPLC003 Adamas 10-2018-LRDokument20 SeitenHPLC003 Adamas 10-2018-LRchiralicNoch keine Bewertungen
- On-Site Fuel and Lubricant Quality Control by Portable FTIR AnalysersDokument2 SeitenOn-Site Fuel and Lubricant Quality Control by Portable FTIR AnalyserschiralicNoch keine Bewertungen
- Lecture 06 - Protective Relay PDFDokument25 SeitenLecture 06 - Protective Relay PDFEmdadul Hoq Rakib100% (1)
- Kubota Front Loader La 211Dokument29 SeitenKubota Front Loader La 211Mark Dubravec40% (5)
- Digital Logic GatesDokument41 SeitenDigital Logic Gatessurafel5248Noch keine Bewertungen
- Math 7-Q4-Module-3Dokument16 SeitenMath 7-Q4-Module-3Jeson GaiteraNoch keine Bewertungen
- Practice Test 3 - Spring 2010Dokument9 SeitenPractice Test 3 - Spring 2010Vasudha97Noch keine Bewertungen
- Understand WorkFlow in DetailDokument118 SeitenUnderstand WorkFlow in DetailSaquib MahmoodNoch keine Bewertungen
- Experiment 6Dokument7 SeitenExperiment 6sajithNoch keine Bewertungen
- Decision Analysis in Quantitative Decision MakingDokument14 SeitenDecision Analysis in Quantitative Decision MakingReader92% (13)
- The 20 Amino Acids and Their Role in Protein Structures: Torsion AnglesDokument3 SeitenThe 20 Amino Acids and Their Role in Protein Structures: Torsion AnglesDaleNoch keine Bewertungen
- 5957-Pan Flute Tone RangesDokument1 Seite5957-Pan Flute Tone Rangesdaniel_gheoldus5867Noch keine Bewertungen
- 4th Quarter Long Quiz in ScienceDokument2 Seiten4th Quarter Long Quiz in ScienceEderlina Bentilanon FagtananNoch keine Bewertungen
- Checking Configuring CPU Blade BIOS SettingsDokument8 SeitenChecking Configuring CPU Blade BIOS SettingsRT05 RW05Noch keine Bewertungen
- Protect transformers from over excitation with relay coordinationDokument7 SeitenProtect transformers from over excitation with relay coordinationAnonymous zxtBoTT100% (1)
- Weekly Home Learning Plan - KindergartenDokument3 SeitenWeekly Home Learning Plan - KindergartenMae Escobin BetonggaNoch keine Bewertungen
- Lab 01 ADokument7 SeitenLab 01 ApathmakerpkNoch keine Bewertungen
- Primo Theory Volume 5 Secured - Unlocked PDFDokument60 SeitenPrimo Theory Volume 5 Secured - Unlocked PDFmsmali100% (1)
- Analyzing Network Traffic with TSharkDokument46 SeitenAnalyzing Network Traffic with TSharkJay SingireddyNoch keine Bewertungen
- Why Python For GISDokument3 SeitenWhy Python For GISalebeley21Noch keine Bewertungen
- WPS For Structure Mild Steel Fillet WeldDokument2 SeitenWPS For Structure Mild Steel Fillet WeldHarkesh Rajput76% (17)
- Electron Configurations and PropertiesDokument28 SeitenElectron Configurations and PropertiesAinthu IbrahymNoch keine Bewertungen
- 505s PDFDokument20 Seiten505s PDFArfan AliNoch keine Bewertungen
- EBS e PDFDokument92 SeitenEBS e PDFRowan Cornelius100% (2)
- EEE312 - Midterm Exam - Part 1 - Google FormsDokument4 SeitenEEE312 - Midterm Exam - Part 1 - Google FormsDavid Louie BediaNoch keine Bewertungen
- Laser Theodolite ManualDokument24 SeitenLaser Theodolite Manualp2342Noch keine Bewertungen
- Thermal Performance AnalysisDokument23 SeitenThermal Performance AnalysisLiew Yau WeiNoch keine Bewertungen
- Steering GeometryDokument23 SeitenSteering GeometryChetanPrasadSevana100% (1)
- Helios PDF HandshakeDokument2 SeitenHelios PDF HandshakemilivojNoch keine Bewertungen
- Antipyretic Ectraction From PlantDokument5 SeitenAntipyretic Ectraction From PlantTaufiksyaefulmalikNoch keine Bewertungen
- GIS Approach To Fire Station Location PDFDokument6 SeitenGIS Approach To Fire Station Location PDFafiNoch keine Bewertungen
- IUPAC Naming by Aravind AroraDokument30 SeitenIUPAC Naming by Aravind Aroratanish gehlotNoch keine Bewertungen