Das könnte Ihnen auch gefallen
- Dry Powder Coating PDFDokument1 SeiteDry Powder Coating PDFMaulik PatelNoch keine Bewertungen
- Plan of WorkDokument3 SeitenPlan of WorkMaulik PatelNoch keine Bewertungen
- Feasibility StudyDokument2 SeitenFeasibility StudyMaulik PatelNoch keine Bewertungen
- Table 1: Composition of Ethyl Cellulose Microspheres SN Ingredient F1 F2 F3 F4 F5 F6Dokument2 SeitenTable 1: Composition of Ethyl Cellulose Microspheres SN Ingredient F1 F2 F3 F4 F5 F6Maulik PatelNoch keine Bewertungen
- List of FigureDokument1 SeiteList of FigureMaulik PatelNoch keine Bewertungen
- Mohit FrontpageDokument1 SeiteMohit FrontpageMaulik PatelNoch keine Bewertungen
- The Yellow House: A Memoir (2019 National Book Award Winner)Von EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Bewertung: 4 von 5 Sternen4/5 (98)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeVon EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeBewertung: 4 von 5 Sternen4/5 (5795)
- Shoe Dog: A Memoir by the Creator of NikeVon EverandShoe Dog: A Memoir by the Creator of NikeBewertung: 4.5 von 5 Sternen4.5/5 (537)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureVon EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureBewertung: 4.5 von 5 Sternen4.5/5 (474)
- Grit: The Power of Passion and PerseveranceVon EverandGrit: The Power of Passion and PerseveranceBewertung: 4 von 5 Sternen4/5 (588)
- On Fire: The (Burning) Case for a Green New DealVon EverandOn Fire: The (Burning) Case for a Green New DealBewertung: 4 von 5 Sternen4/5 (74)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryVon EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryBewertung: 3.5 von 5 Sternen3.5/5 (231)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceVon EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceBewertung: 4 von 5 Sternen4/5 (895)
- Never Split the Difference: Negotiating As If Your Life Depended On ItVon EverandNever Split the Difference: Negotiating As If Your Life Depended On ItBewertung: 4.5 von 5 Sternen4.5/5 (838)
- The Little Book of Hygge: Danish Secrets to Happy LivingVon EverandThe Little Book of Hygge: Danish Secrets to Happy LivingBewertung: 3.5 von 5 Sternen3.5/5 (400)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersVon EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersBewertung: 4.5 von 5 Sternen4.5/5 (345)
- The Unwinding: An Inner History of the New AmericaVon EverandThe Unwinding: An Inner History of the New AmericaBewertung: 4 von 5 Sternen4/5 (45)
- Team of Rivals: The Political Genius of Abraham LincolnVon EverandTeam of Rivals: The Political Genius of Abraham LincolnBewertung: 4.5 von 5 Sternen4.5/5 (234)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyVon EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyBewertung: 3.5 von 5 Sternen3.5/5 (2259)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaVon EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaBewertung: 4.5 von 5 Sternen4.5/5 (266)
- The Emperor of All Maladies: A Biography of CancerVon EverandThe Emperor of All Maladies: A Biography of CancerBewertung: 4.5 von 5 Sternen4.5/5 (271)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreVon EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreBewertung: 4 von 5 Sternen4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Von EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Bewertung: 4.5 von 5 Sternen4.5/5 (121)
- Her Body and Other Parties: StoriesVon EverandHer Body and Other Parties: StoriesBewertung: 4 von 5 Sternen4/5 (821)
- DataStage Interview QuestionsDokument3 SeitenDataStage Interview QuestionsvrkesariNoch keine Bewertungen
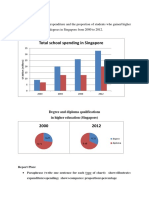
- Total School Spending in Singapore: Task 1 020219Dokument6 SeitenTotal School Spending in Singapore: Task 1 020219viper_4554Noch keine Bewertungen
- Critical Point of View: A Wikipedia ReaderDokument195 SeitenCritical Point of View: A Wikipedia Readerrll307Noch keine Bewertungen
- HS ĐỀ CƯƠNG KIỂM TRA GIỮA KỲ I KHOI 10 NH 23 24Dokument6 SeitenHS ĐỀ CƯƠNG KIỂM TRA GIỮA KỲ I KHOI 10 NH 23 24Ngô Hiểu NhiênNoch keine Bewertungen
- 500 Grammar Based Conversation Question12Dokument16 Seiten500 Grammar Based Conversation Question12Ivo Barry Ward100% (1)
- #Stopableism: Reduksi Stigma Kepada Penyandang Disabilitas Melalui Intervensi Bias ImplisitDokument15 Seiten#Stopableism: Reduksi Stigma Kepada Penyandang Disabilitas Melalui Intervensi Bias ImplisitDennis SuhardiniNoch keine Bewertungen
- ESM Module1 GE3 TCWDokument12 SeitenESM Module1 GE3 TCWDeserie Aguilar DugangNoch keine Bewertungen
- Career Tree EssayDokument2 SeitenCareer Tree Essayapi-437674439Noch keine Bewertungen
- Leasys v1Dokument2 SeitenLeasys v1Arigbede MosesNoch keine Bewertungen
- HiPerMat 2012 KasselDokument1.059 SeitenHiPerMat 2012 KasselAna Mafalda MatosNoch keine Bewertungen
- 27 EtdsDokument29 Seiten27 EtdsSuhag PatelNoch keine Bewertungen
- 1-Employees Retention Strategies in Reliance Retail LTD Thesis 85pDokument6 Seiten1-Employees Retention Strategies in Reliance Retail LTD Thesis 85pArunKumar100% (1)
- ббббббббббббDokument7 Seitenббббббббббббhug1515Noch keine Bewertungen
- D027B OmronDokument141 SeitenD027B OmronirfanWPKNoch keine Bewertungen
- Herzlich Willkommen - Welcome: German Information Session For New ParentsDokument20 SeitenHerzlich Willkommen - Welcome: German Information Session For New ParentssammlissNoch keine Bewertungen
- Proposal Defense Form Chapter 1 3Dokument3 SeitenProposal Defense Form Chapter 1 3Jason BinondoNoch keine Bewertungen
- Stepping Stones Grade 4 ScopeDokument2 SeitenStepping Stones Grade 4 ScopeGretchen BensonNoch keine Bewertungen
- DSpace Configuration PDFDokument1 SeiteDSpace Configuration PDFArshad SanwalNoch keine Bewertungen
- PM Risk For DummiesDokument52 SeitenPM Risk For DummiesFelix Rinaldi100% (8)
- Vertical Axis Wind Turbine ProjDokument2 SeitenVertical Axis Wind Turbine Projmacsan sanchezNoch keine Bewertungen
- Saint Albert Polytechnic College, Inc. Bachelor of Science in Office AdministrationDokument15 SeitenSaint Albert Polytechnic College, Inc. Bachelor of Science in Office AdministrationAustin Grey Pineda MacoteNoch keine Bewertungen
- Normality in The Residual - Hossain Academy NoteDokument2 SeitenNormality in The Residual - Hossain Academy NoteAbdullah KhatibNoch keine Bewertungen
- TELEMAC Code InstructionsDokument3 SeitenTELEMAC Code Instructionskim lee pooNoch keine Bewertungen
- IFEM Ch01Dokument18 SeitenIFEM Ch01Eng AlwardiNoch keine Bewertungen
- Chris Barnes UEFA Presentation Nov 2015Dokument47 SeitenChris Barnes UEFA Presentation Nov 2015muller.patrik1Noch keine Bewertungen
- Job Stress, Workload, Environment and Employee Turnover IntentionsDokument13 SeitenJob Stress, Workload, Environment and Employee Turnover IntentionsYuni SasieNoch keine Bewertungen
- BharathDokument2 SeitenBharathbharath kumarNoch keine Bewertungen
- Kaizen Idea - Sheet: Suitable Chairs To Write Their Notes Faster & Easy Method While TrainingDokument1 SeiteKaizen Idea - Sheet: Suitable Chairs To Write Their Notes Faster & Easy Method While TrainingWahid AkramNoch keine Bewertungen
- Morning Report: Supervisor: Dr. H. Doddy Ak., Spog (K)Dokument10 SeitenMorning Report: Supervisor: Dr. H. Doddy Ak., Spog (K)vika handayaniNoch keine Bewertungen
- Naeyc StandardsDokument11 SeitenNaeyc Standardsapi-266968546Noch keine Bewertungen