Das könnte Ihnen auch gefallen
- Shoe Dog: A Memoir by the Creator of NikeVon EverandShoe Dog: A Memoir by the Creator of NikeBewertung: 4.5 von 5 Sternen4.5/5 (537)
- The Yellow House: A Memoir (2019 National Book Award Winner)Von EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Bewertung: 4 von 5 Sternen4/5 (98)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeVon EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeBewertung: 4 von 5 Sternen4/5 (5794)
- The Little Book of Hygge: Danish Secrets to Happy LivingVon EverandThe Little Book of Hygge: Danish Secrets to Happy LivingBewertung: 3.5 von 5 Sternen3.5/5 (400)
- Grit: The Power of Passion and PerseveranceVon EverandGrit: The Power of Passion and PerseveranceBewertung: 4 von 5 Sternen4/5 (588)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureVon EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureBewertung: 4.5 von 5 Sternen4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryVon EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryBewertung: 3.5 von 5 Sternen3.5/5 (231)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceVon EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceBewertung: 4 von 5 Sternen4/5 (895)
- Team of Rivals: The Political Genius of Abraham LincolnVon EverandTeam of Rivals: The Political Genius of Abraham LincolnBewertung: 4.5 von 5 Sternen4.5/5 (234)
- Never Split the Difference: Negotiating As If Your Life Depended On ItVon EverandNever Split the Difference: Negotiating As If Your Life Depended On ItBewertung: 4.5 von 5 Sternen4.5/5 (838)
- The Emperor of All Maladies: A Biography of CancerVon EverandThe Emperor of All Maladies: A Biography of CancerBewertung: 4.5 von 5 Sternen4.5/5 (271)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaVon EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaBewertung: 4.5 von 5 Sternen4.5/5 (266)
- On Fire: The (Burning) Case for a Green New DealVon EverandOn Fire: The (Burning) Case for a Green New DealBewertung: 4 von 5 Sternen4/5 (74)
- The Unwinding: An Inner History of the New AmericaVon EverandThe Unwinding: An Inner History of the New AmericaBewertung: 4 von 5 Sternen4/5 (45)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersVon EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersBewertung: 4.5 von 5 Sternen4.5/5 (345)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyVon EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyBewertung: 3.5 von 5 Sternen3.5/5 (2259)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreVon EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreBewertung: 4 von 5 Sternen4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Von EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Bewertung: 4.5 von 5 Sternen4.5/5 (121)
- Her Body and Other Parties: StoriesVon EverandHer Body and Other Parties: StoriesBewertung: 4 von 5 Sternen4/5 (821)
- Foly Catheter Removal ProtocolDokument1 SeiteFoly Catheter Removal ProtocolMostafa ShamaaNoch keine Bewertungen
- Preface: Encyclopedia (Marshall Sittig, Noyes Publications, WestwoodDokument4 SeitenPreface: Encyclopedia (Marshall Sittig, Noyes Publications, WestwoodJulcon Avanceña AraizNoch keine Bewertungen
- Sanofi PriceListDokument1 SeiteSanofi PriceListJohn EnochNoch keine Bewertungen
- A Review On Extended Release Drug Delivery SystemDokument9 SeitenA Review On Extended Release Drug Delivery SystemTuyến Đặng ThịNoch keine Bewertungen
- ACECLOFENACDokument7 SeitenACECLOFENACShifanath ANoch keine Bewertungen
- The Efficacy of Platelet-Rich Plasma in The Field of Hair Restoration and Facial Aesthetics A Systematic Review and MetaanalysisDokument19 SeitenThe Efficacy of Platelet-Rich Plasma in The Field of Hair Restoration and Facial Aesthetics A Systematic Review and MetaanalysisMariska Regina RannuNoch keine Bewertungen
- Top Prescription DrugsDokument3 SeitenTop Prescription DrugsArturo MacaranasNoch keine Bewertungen
- CCO Invasive Fungal Disease Downloadable 1Dokument25 SeitenCCO Invasive Fungal Disease Downloadable 1Vasantha KumarNoch keine Bewertungen
- Degroat RN ResumeDokument1 SeiteDegroat RN Resumeapi-363089889Noch keine Bewertungen
- OPPOSITE: Legalization of Marijuana in The PhilippinesDokument2 SeitenOPPOSITE: Legalization of Marijuana in The PhilippinesNathalie GetinoNoch keine Bewertungen
- EP2477611B1Dokument2 SeitenEP2477611B1Yahya RizkiNoch keine Bewertungen
- Assessment Pada Ujian OSCEDokument38 SeitenAssessment Pada Ujian OSCEAnggraeni KusumaratihNoch keine Bewertungen
- Alternative MedicineDokument3 SeitenAlternative MedicineAntoniaMottaNoch keine Bewertungen
- MSD Numbers 0410612020 Am: ClosingDokument11 SeitenMSD Numbers 0410612020 Am: ClosingSanjeev JayaratnaNoch keine Bewertungen
- Biology Project: TOPIC - Study of Drugs, Their Types and Alcohol Abuse To AdolescentsDokument13 SeitenBiology Project: TOPIC - Study of Drugs, Their Types and Alcohol Abuse To AdolescentsSAFDAR HafizNoch keine Bewertungen
- Basics of Pharmacology: Speaker - DR - SantoshDokument76 SeitenBasics of Pharmacology: Speaker - DR - SantoshRamya ChallaNoch keine Bewertungen
- Dr. Rathnakar U.P.: Department of Pharmacology Kasturba Medical College, MangaloreDokument25 SeitenDr. Rathnakar U.P.: Department of Pharmacology Kasturba Medical College, MangaloreDr.U.P.Rathnakar.MD.DIH.PGDHM100% (1)
- Chemotherapeutic DrugsDokument4 SeitenChemotherapeutic DrugsEditor IJTSRDNoch keine Bewertungen
- Laporan Penggunaan Maret 2020Dokument6 SeitenLaporan Penggunaan Maret 2020siska thresiaNoch keine Bewertungen
- Daftar Obat Aman Dan Berbahaya Untuk Ibu Hamil DanDokument6 SeitenDaftar Obat Aman Dan Berbahaya Untuk Ibu Hamil Danirma suwandi sadikinNoch keine Bewertungen
- Top 200 DrugsDokument4 SeitenTop 200 DrugsEsther AhnNoch keine Bewertungen
- Drug and ClassificationDokument4 SeitenDrug and ClassificationdavidcalaloNoch keine Bewertungen
- Nsaids BcqsDokument1 SeiteNsaids BcqsDR AbidNoch keine Bewertungen
- Penggunaan Obat-Obat KhususDokument39 SeitenPenggunaan Obat-Obat KhususDeisy FebriantiNoch keine Bewertungen
- Fibromyalgia: by Tara E. Dymon, Pharm.D., BCACPDokument14 SeitenFibromyalgia: by Tara E. Dymon, Pharm.D., BCACPAnggi CalapiNoch keine Bewertungen
- Pep 2021 - October QuestionsDokument4 SeitenPep 2021 - October QuestionsCynthia ObiNoch keine Bewertungen
- Daftar Ketersediaan Obat Sesuai Formularium Nasional Puskesmas Candi TAHUN 2020Dokument18 SeitenDaftar Ketersediaan Obat Sesuai Formularium Nasional Puskesmas Candi TAHUN 2020octie0% (1)
- Pharmacovigilance Related Questions - 1585456120320Dokument10 SeitenPharmacovigilance Related Questions - 1585456120320Saravanan RamNoch keine Bewertungen
- Pharma MCQ214324324325Dokument17 SeitenPharma MCQ214324324325rab yoNoch keine Bewertungen
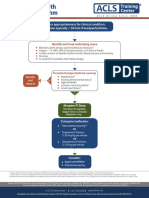
- Assess Appropriateness For Clinical Condition. Heart Rate Typically 50/min If BradyarrhythmiaDokument1 SeiteAssess Appropriateness For Clinical Condition. Heart Rate Typically 50/min If BradyarrhythmiaatikaNoch keine Bewertungen