Das könnte Ihnen auch gefallen
- The Yellow House: A Memoir (2019 National Book Award Winner)Von EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Bewertung: 4 von 5 Sternen4/5 (98)
- Easter Island or Rapa NuiDokument6 SeitenEaster Island or Rapa NuiSofía I. Morales NavarroNoch keine Bewertungen
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceVon EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceBewertung: 4 von 5 Sternen4/5 (895)
- A Framework For The Integration of Ecosystem and Human HealthDokument14 SeitenA Framework For The Integration of Ecosystem and Human HealthSofía I. Morales NavarroNoch keine Bewertungen
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeVon EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeBewertung: 4 von 5 Sternen4/5 (5794)
- Brain Structure SummaryDokument2 SeitenBrain Structure SummarySofía I. Morales NavarroNoch keine Bewertungen
- The Little Book of Hygge: Danish Secrets to Happy LivingVon EverandThe Little Book of Hygge: Danish Secrets to Happy LivingBewertung: 3.5 von 5 Sternen3.5/5 (399)
- Urban Ecology and Urban Ecosystems, Understanding The Links To Human Health and Well BeingDokument8 SeitenUrban Ecology and Urban Ecosystems, Understanding The Links To Human Health and Well BeingSofía I. Morales NavarroNoch keine Bewertungen
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaVon EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaBewertung: 4.5 von 5 Sternen4.5/5 (266)
- Solubility Rules Made EasyDokument1 SeiteSolubility Rules Made EasySofía I. Morales NavarroNoch keine Bewertungen
- Shoe Dog: A Memoir by the Creator of NikeVon EverandShoe Dog: A Memoir by the Creator of NikeBewertung: 4.5 von 5 Sternen4.5/5 (537)
- A Framework For The Integration of Ecosystem and Human HealthDokument14 SeitenA Framework For The Integration of Ecosystem and Human HealthSofía I. Morales NavarroNoch keine Bewertungen
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureVon EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureBewertung: 4.5 von 5 Sternen4.5/5 (474)
- Interview QuestionsDokument1 SeiteInterview QuestionsSofía I. Morales NavarroNoch keine Bewertungen
- Never Split the Difference: Negotiating As If Your Life Depended On ItVon EverandNever Split the Difference: Negotiating As If Your Life Depended On ItBewertung: 4.5 von 5 Sternen4.5/5 (838)
- Interview QuestionsDokument1 SeiteInterview QuestionsSofía I. Morales NavarroNoch keine Bewertungen
- Grit: The Power of Passion and PerseveranceVon EverandGrit: The Power of Passion and PerseveranceBewertung: 4 von 5 Sternen4/5 (588)
- Med School Interview QuestionsDokument4 SeitenMed School Interview QuestionsSofía I. Morales NavarroNoch keine Bewertungen
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryVon EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryBewertung: 3.5 von 5 Sternen3.5/5 (231)
- Amphetamines: ProfileDokument5 SeitenAmphetamines: ProfileSofía I. Morales NavarroNoch keine Bewertungen
- Med School Interview QuestionsDokument4 SeitenMed School Interview QuestionsSofía I. Morales NavarroNoch keine Bewertungen
- The Emperor of All Maladies: A Biography of CancerVon EverandThe Emperor of All Maladies: A Biography of CancerBewertung: 4.5 von 5 Sternen4.5/5 (271)
- Table of Physical ConstantsDokument1 SeiteTable of Physical ConstantsSofía I. Morales NavarroNoch keine Bewertungen
- HFCS Everything You Wanted To Know But Were Afraid To AskDokument1 SeiteHFCS Everything You Wanted To Know But Were Afraid To AskSofía I. Morales NavarroNoch keine Bewertungen
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyVon EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyBewertung: 3.5 von 5 Sternen3.5/5 (2259)
- The Effects of HFCSDokument8 SeitenThe Effects of HFCSSofía I. Morales NavarroNoch keine Bewertungen
- On Fire: The (Burning) Case for a Green New DealVon EverandOn Fire: The (Burning) Case for a Green New DealBewertung: 4 von 5 Sternen4/5 (73)
- Chapter 1 Introduction To Emergency Medical CareDokument38 SeitenChapter 1 Introduction To Emergency Medical CareSofía I. Morales NavarroNoch keine Bewertungen
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersVon EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersBewertung: 4.5 von 5 Sternen4.5/5 (344)
- Nmat Reviewer Orgchem PDFDokument15 SeitenNmat Reviewer Orgchem PDFAlice Katrina100% (1)
- Chapter 2 COE Alkenes and Alkynes II Addition ReactionsDokument54 SeitenChapter 2 COE Alkenes and Alkynes II Addition ReactionsRaven ShadeNoch keine Bewertungen
- Team of Rivals: The Political Genius of Abraham LincolnVon EverandTeam of Rivals: The Political Genius of Abraham LincolnBewertung: 4.5 von 5 Sternen4.5/5 (234)
- Chapter 6: Reactions of Alkenes - Addition Reactions: Trans-1,2-Dimethylcyclopentane Cis-1,2-DimethylcyclopentaneDokument21 SeitenChapter 6: Reactions of Alkenes - Addition Reactions: Trans-1,2-Dimethylcyclopentane Cis-1,2-DimethylcyclopentaneRahma AshrafNoch keine Bewertungen
- @iitjeeadvancedmaterial Alcohols, Phenols & EthersDokument148 Seiten@iitjeeadvancedmaterial Alcohols, Phenols & EthersRonak kadamNoch keine Bewertungen
- Hydrocarbon-04 Solved ProblemsDokument14 SeitenHydrocarbon-04 Solved ProblemsRaju SinghNoch keine Bewertungen
- The Unwinding: An Inner History of the New AmericaVon EverandThe Unwinding: An Inner History of the New AmericaBewertung: 4 von 5 Sternen4/5 (45)
- Haloalkanes and HaloarenesDokument28 SeitenHaloalkanes and HaloarenesDevansh TiwaryNoch keine Bewertungen
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreVon EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreBewertung: 4 von 5 Sternen4/5 (1090)
- Alkynes: An Introduction To Organic Synthesis: (Ref: Mcmurry'S Organic Chemistry, 7 Edition)Dokument24 SeitenAlkynes: An Introduction To Organic Synthesis: (Ref: Mcmurry'S Organic Chemistry, 7 Edition)Tristan BadillaNoch keine Bewertungen
- 27 Alcohol Phenol Ether Formula Sheets Getmarks AppDokument15 Seiten27 Alcohol Phenol Ether Formula Sheets Getmarks AppFLASH FFNoch keine Bewertungen
- CHE 112 - Lecture 2Dokument103 SeitenCHE 112 - Lecture 2Martias WambiNoch keine Bewertungen
- Alkene Lecture - 3Dokument8 SeitenAlkene Lecture - 3Sukanya PhukonNoch keine Bewertungen
- CBSE Class 12 Chemistry Chapter 10 Haloalkanes and Haloarenes Revision NotesDokument99 SeitenCBSE Class 12 Chemistry Chapter 10 Haloalkanes and Haloarenes Revision NotesTECH STOVENoch keine Bewertungen
- CHM 2210 Practice Exam 3Dokument8 SeitenCHM 2210 Practice Exam 3Shaima MossamatNoch keine Bewertungen
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Von EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Bewertung: 4.5 von 5 Sternen4.5/5 (121)
- Organic Chemistry 2 Practice Exam 1Dokument15 SeitenOrganic Chemistry 2 Practice Exam 1KaybidoNoch keine Bewertungen
- Reaction of AlkenesDokument27 SeitenReaction of AlkenesHui JingNoch keine Bewertungen
- Mechanism of Organic ReactionsDokument45 SeitenMechanism of Organic ReactionsBapu ThoratNoch keine Bewertungen
- JPP-5 - (JLD 3.0) - Reaction Mechanisms - 05 SeptDokument50 SeitenJPP-5 - (JLD 3.0) - Reaction Mechanisms - 05 SeptHarsh ThakurNoch keine Bewertungen
- Chapter 9 Orgo Chem Test Bank PDFDokument77 SeitenChapter 9 Orgo Chem Test Bank PDFahmshiNoch keine Bewertungen
- Team Bootcamp - Reaction Summary Sheet PDFDokument30 SeitenTeam Bootcamp - Reaction Summary Sheet PDFDagne PovedaNoch keine Bewertungen
- Free Radical SubstitutionDokument22 SeitenFree Radical SubstitutionULFA TUFFAHATI100% (1)
- Reaction Road MapDokument2 SeitenReaction Road MapBenedick Jayson MartiNoch keine Bewertungen
- Haloalkanes And: HaloarenesDokument24 SeitenHaloalkanes And: HaloarenesWild SageNoch keine Bewertungen
- Chapter 3 AlkenesDokument63 SeitenChapter 3 AlkenesKonoli NuingNoch keine Bewertungen
- Overview of Alkene ReactionsDokument11 SeitenOverview of Alkene ReactionsVickyNoch keine Bewertungen
- Her Body and Other Parties: StoriesVon EverandHer Body and Other Parties: StoriesBewertung: 4 von 5 Sternen4/5 (821)
- 5.2 Alkene 2Dokument6 Seiten5.2 Alkene 2Yusra IqbalNoch keine Bewertungen
- Reaction Mechanisms OverviewDokument19 SeitenReaction Mechanisms OverviewHanhHongDaoNoch keine Bewertungen
- Chapter 10 Alkenes - SmithDokument14 SeitenChapter 10 Alkenes - SmithRen Liew Jia Qing75% (4)
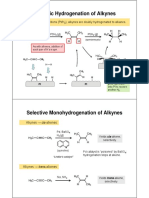
- Catalytic Hydrogenation of Alkynes: As With Alkenes, Addition of Each Pair of H's Is SynDokument6 SeitenCatalytic Hydrogenation of Alkynes: As With Alkenes, Addition of Each Pair of H's Is SynVishal SinghNoch keine Bewertungen
- Organic ChemistryDokument20 SeitenOrganic ChemistryGirish RaguvirNoch keine Bewertungen
- L14 - (JLD 3.0) - Reaction Mechanisms - 02 SeptDokument62 SeitenL14 - (JLD 3.0) - Reaction Mechanisms - 02 SeptkisuisoffbeatNoch keine Bewertungen
- Oc 1. Alkynes and Alkadienes Final RK Sir - 05.03.14 (01-16) PDFDokument16 SeitenOc 1. Alkynes and Alkadienes Final RK Sir - 05.03.14 (01-16) PDFAman9692100% (1)