Beruflich Dokumente
Kultur Dokumente
Tese Ismael 040111
Hochgeladen von
Ismael LeiteCopyright
Verfügbare Formate
Dieses Dokument teilen
Dokument teilen oder einbetten
Stufen Sie dieses Dokument als nützlich ein?
Sind diese Inhalte unangemessen?
Dieses Dokument meldenCopyright:
Verfügbare Formate
Tese Ismael 040111
Hochgeladen von
Ismael LeiteCopyright:
Verfügbare Formate
UNIVERSIDADE FEDERAL DO CEAR FACULDADE DE MEDICINA DEPARTAMENTO DE FISIOLOGIA E FARMACOLOGIA PROGRAMA DE PS-GRADUAO EM FARMACOLOGIA
ISMAEL LEITE MARTINS
DESENVOLVIMENTO E VALIDAO DE MTODOS BIOANALTICOS PARA QUANTIFICAO DA AMOXICILINA, NORFLOXACINO E OXCARBAZEPINA EM ESTUDOS FARMACOCINTICOS
FORTALEZA 2009
ISMAEL LEITE MARTINS
DESENVOLVIMENTO E VALIDAO DE MTODOS BIOANALTICOS PARA QUANTIFICAO DA AMOXICILINA, NORFLOXACINO E OXCARBAZEPINA EM ESTUDOS FARMACOCINTICOS
Tese submetida coordenao do Programa de PsGraduao em Farmacologia, do Departamento de Fisiologia e Farmacologia da Faculdade de Medicina da Universidade Federal do Cear, como requisito parcial para obteno do Grau de Doutor em Farmacologia.
Orientadora: Profa. Dra. Maria Elisabete Amaral de Moraes Co-Orientadora: Profa. Dra. Maria Bernadete de Sousa Maia
FORTALEZA 2009
M343d
Martins, Ismael Leite Desenvolvimento e Validao de Mtodos Bioanalticos para Quantificao da Amoxicilina, Norfloxacino e Oxcarbazepina em Estudos Farmacocinticos/ Ismael Leite Martins. - Fortaleza, 2009. 195 f. : il Orientadora: Profa. Dra. Maria Elisabete Amaral de Moraes Tese (Doutorado) - Universidade Federal do Cear. Departamento de Fisiologia e Farmacologia. Fortaleza, Cear. 1. Farmacocintica 2. Cromatografia 3. Amoxicilina 4. Norfloxacino I. Moraes, Maria Elisabete Amaral de (Orient.). II. Ttulo CDD: 615.7
ISMAEL LEITE MARTINS DESENVOLVIMENTO E VALIDAO DE MTODOS BIOANALTICOS PARA QUANTIFICAO DA AMOXICILINA, NORFLOXACINO E OXCARBAZEPINA EM ESTUDOS FARMACOCINTICOS Tese submetida Coordenao do Programa de Ps-Graduao em Farmacologia da Universidade Federal do Cear, como requisito parcial para obteno do grau de Doutor em Farmacologia. Aprovada em: 29 de julho de 2009. BANCA EXAMINADORA
Dedico esta tese a todas as preciosidades que Deus me deu: meu pai, minha me, minha irm, meu benzim, por todo amor e incentivo em todas as etapas importantes da minha vida.
AGRADECIMENTOS
Deus, pela minha vida repleta de bnos, que hoje possibilitam a concretizao de um sonho. Minha Famlia, pelo carinho, apoio, incentivo em todas as situaes, e pelo entusiasmo conquista. minha futura esposa, Lgia Uchoa, pelo carinho, apoio, amor e companhia constantes e por ter sempre um sorriso a me oferecer. Aos fundadores da Unidade de Farmacologia Clnica, os professores Dra. Maria Elisabete Amaral de Moraes, Dr. Manoel Odorico de Moraes, Dr. Fernando Antonio Frota Bezerra, pela acolhida de todos esses anos, por dividirem comigo seus conhecimentos, por tornar palpvel o sonho da Farmacologia Clnica no Estado do Cear e por to caloroso convvio. professora Dra. Maria Elisabete Amaral de Moraes, minha orientadora, a qual me apoiou na construo do saber cientfico, por depositar incondicionalmente sua confiana no meu trabalho e corrigindo minhas imperfeies. professora Dra. Maria Bernadete Sousa Maia, por colaborar sempre com seus ensinamentos enriquecedores que muito contriburam para o desenvolvimento deste trabalho. professora Dra. Gisela Costa Camaro, pelo grande amor ao ensino e pesquisa, que se reflete em cada palavra proferida e, tambm, pela ateno a mim dispensada. professora Dra. Gilmara Silva de Melo Santana, por compartilhar comigo tantos momentos importantes para meu crescimento pessoal e profissional tornando mais harmoniosa e gratificante a conquista de mais uma etapa em minha vida.
Ao Dr. Vagnaldo Fechine pela ateno peculiar e colaborao na anlise dos dados contidos neste trabalho. Aos Professores do Curso de Ps-Graduao, por terem transmitido com muita sapincia seus conhecimentos. Aos Amigos da Unidade de Farmacologia Clnica, meu agradecimento a todos vocs por terem colaborado na minha pesquisa cientfica, dividindo experincias e amenizando o peso dos trabalhos. Maria Teresa pela colaborao e presteza, bem como pelos momentos agradveis que tivemos. ura, secretria do Departamento de Fisiologia e Farmacologia, que sempre estive disposta a nos ajudar no que fosse preciso. Ao Conselho Nacional de Pesquisa (CNPq), Fundao Cearense de Apoio ao Desenvolvimento Cientfico e Tecnolgico (FUNCAP), Financiadora de Estudos e Projetos (FINEP), DECIT/MS/MCT e Instituto Claude Bernard (InCB), por ter colaborado financeiramente no desenvolvimento da pesquisa. A todos aqueles que de forma direta ou indireta contriburam para a realizao deste trabalho, os meus sinceros agradecimentos.
No com honras e riquezas que se contenta o corao de um pensador, mas sim apenas com o saber e a virtude.
Plato 427 a.C. - id. c. 347 a.C.
RESUMO
DESENVOLVIMENTO E VALIDAO DE MTODOS BIOANALTICOS PARA QUANTIFICAO DA AMOXICILINA, NORFLOXACINO E OXCARBAZEPINA EM ESTUDOS FARMACOCINTICOS. Ismael Leite Martins. Orientadora: Prof. Dra. Maria Elisabete Amaral de Moraes. Tese apresentada ao Programa de PsGraduao em Farmacologia do Departamento de Fisiologia e Farmacologia da Faculdade de Medicina da Universidade Federal do Cear para obteno do Grau de Doutor em Farmacologia. Fortaleza, 2009. Foram desenvolvidos e validados trs mtodos robustos para a determinao de amoxicilina, norfloxacino, oxcarbazepina (OXC) e 10,11-dihidro-10hidroxicarbamazepina (MHD) em plasma, utilizando cromatografia lquida de alta eficincia em fase reversa (RP-HPLC) com deteco ultravioleta, fluorescncia e espectrometria de massa, respectivamente. Os mtodos envolveram extrao lquido-lquido, com diclorometano (amoxicilina), acetonitrila (amoxicilina e norfloxacino), ter etlico-diclorometano (60:40 v/v, oxcarbazepina), utilizando cefadroxil, ciprofloxacino e d10-carbamazepina como padres interno (PI). Separaes cromatogrficas foram realizadas utilizando colunas Gemini C18 5 m (150 X 4,6 mm), Synergi RP-MAX 4 m (150 X 4,6 mm) e Luna C18 m (150 X 4,6 mm), com sistemas de eluio constitudos por mistura de tampo fosfato de 0,01 M (pH 3,5)/acetonitrila (95:5 v/v), acetonitrila-tampo fosfato (85:15, v/v) e acetonitrilagua (50:50 v/v) + 20 mM cido actico, respectivamente. As curvas de calibrao foram lineares, nas faixas de 0,5 a 40 g/mL, 30 a 3500 ng/mL, 20 a 5250 ng/ml e 40 a 10500 ng/ml. As recuperaes nas concentraes de 1,5, 15 e 30 g/ml foram de 59,4%, 60,5% e 67,1% para amoxicilina; 90, 1400 e 2800 ng/mL foram de 103,5%, 100,2% e 100,2% para norfloxacino, 60, 2000 e 4000 ng/ml foram de 105,4, 89,2 e 92,8% para OXC e 120, 4000 e 8000 ng/ml foram de 88,4, 88,7 e 90,6% para MHD, respectivamente. Os mtodos validados incluram avaliao de preciso e exatido intra e interlote, assegurando que estes estavam dentro de limites admissveis. Os mtodos foram ento aplicados com sucesso em estudos de bioequivalncia, administrando 500 mg ou 400 mg das formulaes referncia/teste de amoxicilina e norfloxacino, e em estudos farmacocinticos com formulao de oxcarbazepina suspenso (6%), em voluntrios sadios. Palavras-chave: Farmacocintica. Cromatografia. Amoxicilina. Norfloxacino. Oxcarbazepina.
ABSTRACT DEVELOPMENT AND VALIDATION OF BIOANALYTICAL METHODS FOR QUANTIFICATION OF AMOXICILLIN, NORFLOXACIN AND OXCARBAZEPINE IN PHARMACOKINETIC STUDIES. Ismael Leite Martins. Adviser: Maria Elisabete Amaral de Moraes. Thesis presented for the title of Doctor in Pharmacology. Department of Physiology and Pharmacology, Faculty of Medicine, Federal University of Cear. Fortaleza, 2009. A robust method for the determination of amoxicillin, norfloxacin, oxcarbazepine (OXC) and its active metabolite, 10,11-dihydro-10hydroxycarbamazepine (MHD) in human plasma, using reversed-phase highperformance liquid chromatography (RP-HPLC) with ultraviolet, fluorescence and mass spectrometry detection, respectively, have been developed and valited. The methods involve precipitation of plasma protein with dichloromethane (amoxicillin), acetonitrile (amoxicillin and norfloxacin) and diethyl etherdiclhoromethane (60:40 v/v, oxcarbazepine), using cefadroxil, ciprofloxacin and deuterade carbamazepine (d10-carbamazepine) as internal standard (IS). Chromatographic separations were performed on a column Gemini C18 5 m (150 X 4.6 mm), Synergi MAX-RP 4 m (150 X 4.6 mm) and Luna C18 5 m (150mm X 4.6 mm) with an elution system consisting of a mixture of 0.01 M buffer phosphate (pH 3.5)/acetonitrile (95:05 v/v), phosphate bufferacetonitrile (85:15, v/v) and acetonitrile/water (50:50 v/v) + 20mM acetic acid, respectively. The calibration curve was linear, in the range of 0.5 to 40 g/mL, 30 to 3500 ng/mL, 20 to 5250 ng/mL and 40 to 10,500 ng/mL. The recoveries at concentrations of 1.5, 15 and 30 g/mL were foram 59.4%, 60.5% and 67.1% for amoxicillin; 90, 1400 and 2800 ng/mL were 103.5%, 100.2% and 100.2% for norfloxacin, 60, 2000 and 4000 ng/mL were 105.4, 89.2 and 92.8% for OXC and 120, 4000 and 8000 ng/mL were 88.4, 88.7 and 90.6% for MHD, respectively. The statistical evaluation of the developed method was conducted by examining withinbatch and between-batch precision data, which were within the required limits. The methods were successfully applied in bioequivalence studies given 500 or 400-mg of the reference formulation / test amoxicillin and norfloxacin, and pharmacokinetic studies with formulation of oxcarbazepine suspension (6%) in healthy volunteers. Keywords: Chromatography. Pharmacokinetics. Amoxicillin. Norfloxacin. Oxcarbazepine.
LISTA DE QUADROS 1. 2. 3. 4. 5. Parmetros de validao do Inmetro e Anvisa ........................................ Parmetros de conformidade do sistema e recomendaes .................. Parmetros utilizados na validao do mtodo ....................................... Parmetros Farmacocinticos do Norfloxacino ....................................... Parmetros Farmacocinticos do Oxcarbazepina e MHD ...................... 26 51 82 87 89
LISTA DE TABELAS 1. 2. 3. Utenslios utilizados no desenvolvimento e validao dos mtodos. Validao da quantificao de amoxicilina em plasma: preciso intra e interlote das amostras de controle de qualidade (n = 8) ......................... Resultado da estabilidade (mdia desvio padro) obtido com trs amostras de plasma com concentrao baixa (1,5 g/mL), mdia (15 g/mL) e alta (30 g/mL) de amoxicilina ................................................. 4. 5 6. 7. 8. Parmetros farmacocinticos da formulao teste e referncia (amoxicilina 500 mg) depois da administrao oral de 24 voluntrios .... Mdia, IC (90%) e concluso para a razo das mdias de Cmax, AUC0ult h
106 130
131 132 133
e AUC0- Dados transformados em logaritmo natural.
Dados de preciso e exatido intra e interlote para o norfloxacino para validao em plasma ............................................................................... 140 Recuperao do norfloxacino e ciprofloxacino em plasma ..................... Teste de estabilidade (teste de estabilidade ps-processamento, estabilidade congelamento e descongelamento, estabilidade curta 15 horas e longa durao 44 dias). Nmero de replicatas: n = 5 ................. 142 141
9.
Resultados
das
anlises
de
bioequivalncia
para
relao 144 152
teste/referncia depois da transformao logartmica dos parmetros AUC0-, AUC (0-24h) e Cmax......................................................................... 10. 11. Preciso e exatido intralote das curvas de calibrao........................... Preciso e exatido na quantificao de OXC e MHD em amostras de plasma (anlise de amostras de plasma contaminado em quatro concentraes diferentes) ....................................................................... 12. Estabilidade das amostras de controle de qualidade da OXC e MHD em plasma sob diferentes condies de estoque (n=5) .......................... 156 153
LISTA DE FIGURAS
01.
Determinao grfica das curvas de linearidade atravs da: (a) curva analtica clssica; (b) grfico da razo sinal/concentrao vs. concentrao em escala logartmica..................................................... 32
02.
Modos de diluio da soluo estoque: a) diluio a partir de uma soluo estoque e b) diluio a partir de duas solues estoques (1 e 2) preparadas com concentraes diferentes....................................... 37
03.
Aparelho de cromatografia lquida de alta eficincia com deteco de ultravioleta utilizado na quantificao das amostras contendo amoxicilina. 56
04.
Aparelho de cromatografia lquida de alta eficincia com deteco de fluorescncia utilizado na quantificao das amostras contendo norfloxacino. 57 58
05. 06.
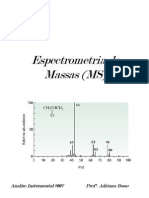
Esquema de funcionamento do sistema de espectrometria de massa Aparelho de cromatografia lquida de alta eficincia acoplado ao espectrmetro de massa utilizado na quantificao das amostras contendo oxcarbazepina.
59 triplo-quadrupolo no modo de 60 62 63 65 102 112 113
07.
Esquema demonstrativo da fragmentao de compostos utilizando espectrmetro de massa monitoramento de reao mltipla (MRM) ...........................................
08. 09. 10. 11. 12. 13. 14.
Processo de absoro de formas farmacuticas slidas ..................... Distribuio de frmacos no organismo ............................................... Biotransformao de frmacos ............................................................. Esquema do procedimento de coleta de sangue e separao de amostras plasmticas. Esquema dos ensaios de estabilidade em ciclos de congelamento de descongelamento Esquema dos ensaios de estabilidade de longa durao. Cromatograma representativo do plasma branco (A), plasma contaminado com amoxicilina (30 g/mL) e cefadroxil (padro interno) (B) e plasma de um voluntrio aps 1h da administrao oral de amoxicilina .......................................................................................
128
15 16.
Representao grfica da curva de calibrao y = 19,0642x + 0,000162149, r2= 0,999253.................................................................. Grfico da curva de concentrao plasmtica (g/dL) versus tempo (h) da amoxicilina. Representa a mdia das concentraes plasmticas obtidas aps a administrao de duas formulaes de Amoxicilina 500 mg em volutrios sadios.............................................. 133 129
17.
Cromatografia de amostra de plasma branco extrado (A) e de plasma contaminado contendo norfloxacino (90 ng/mL) e padro interno (B) ............................................................................................. 138 139
18 19.
Representao grfica da curva de calibrao y = 985,11x 2,52151, r2= 0,999493.......................................................................................... Curva de concentrao plasmtica mdia versus tempo aps administrao de duas formulaes de norfloxacino 400 mg em voluntrios sadios ................................................................................. 143 148
20. 21.
Full-scan dos espectros dos ons fragmento de [M+H]+ e as estruturas da (a) OXC, (b) MHD e (c) d10-carbamazepina .................. Cromatogramas MRM: (a) uma amostra de plasma em branco, (b) uma amostra de plasma branco contaminada com OXC (20 ng / ml) e MHD (40 ng / ml), (c) um voluntrio amostra 1,5 h aps a administrao oral de OXC suspenso (6%) em voluntrios saudveis. Picos I-III referem-se ao MHD, OXC e PI, 150 respectivamente ...................................................................................
22.
Perfil da curva de concentrao plasmtica mdia de OXC e MHD versus tempo aps administrao de 10 ml de uma suspenso oral de oxcarbazepina (6%) em voluntrio sadios (n=8) ............................. 157
LISTA DE ABREVIATURAS E SIGLAS % < m L
o
Porcentagem Mais ou Menos Menor que Menor ou igual a Micrometro Microlitro Grau Celsius Anlise de Varincia Agncia Nacional de Vigilncia Sanitria Conselho Nacional de Sade Comit de tica em Pesquisa da UFC Coeficiente de Variao Desvio Padro Grama Grama por Decilitro Litro Miligrama Minuto Mililitro Milmetro Ministrio da Sade Nanmetro Nmero Organizao Mundial da Sade Universidade Federal do Cear Area Under Curve Collisionally Activated Dissociation Collision-Induced Dissociation High Performance/Pressure Liquide Chromatography Concentrao Mxima Controle de Qualidade Controle de Qualidade Alto
ANOVA Anvisa CNS COMEPE CV DP g g/dL L mg Min mL mm MS nm N OMS UFC AUC CAD CID HPLC Cmax CQ CQA
CQB CQM FDA ICH IES IMC ISO Ke LQ MRM MS/MS t1/2 UNIFAC USP
Controle de Qualidade Baixo Controle de Qualidade Mdio Food and Drug Administration International Conference on Harmonisation Ionizao por Eletrospray ndice de Massa Corporal Internacional Standard Organization Constante de eliminao Limite de Quantificao Monitoramento de reao mltipla Mass spectrometry Tempo da meia-vida plasmtica Unidade de Farmacologia Clnica United States Pharmacopoeia
SUMRIO PARTE I: INTRODUO E REVISO BIBLIOGRFICA CAPTULO 1: Validao de Mtodos Bioanalticos 1 INTRODUO 2 CONCEITOS BSICOS DO PROCESSO DE VALIDAO 3 LEGISLAO 4 PROCESSO DE VALIDAO 5 PARMETROS ANALTICOS PARA VALIDAO DE MTODOS 5.1 Seletividade 5.2 Linearidade e Faixa de Aplicao 5.2.1 Padronizao externa 5.2.2 Padronizao interna 5.2.3 Superposio de matriz 5.2.4 Adio padro 5.3 Preciso 5.3.1 Repetitividade 5.3.2 Preciso intermediria 5.3.3 Reprodutibilidade 5.4 Exatido 5.4.1 Materiais de referncia certificados (CRM) 5.4.2 Comparao de mtodos 5.4.3 Ensaios de recuperao 5.4.4 Adio padro 5.5 Limite de Deteco (LD) 5.5.1 Mtodo visual 5.5.2 Mtodo da relao sinal-rudo 5.5.3 Mtodo baseado em parmetros da curva analtica 5.6 Limite de Quantificao (LQ) 5.7 Robustez 6 ESTABILIDADE DOS PADRES E DAS AMOSTRAS 7 CONFORMIDADE DO SISTEMA 8 REVALIDAO CAPTULO 2: Sistema de Deteco de Frmacos 22 23 23 23 25 27 28 28 30 33 33 34 35 37 38 39 39 40 41 42 43 44 45 45 45 46 46 47 49 49 51 52
1 CROMATOGRAFIA LQUIDA DE ALTA EFICINCIA (HPLC) 1.1 Detector por Absorbncia no Ultravioleta e no Visvel 1.2 Detector por Fluorescncia 1.3 Detector por Espectrometria de Massa CAPTULO 3: Estudos Farmacocinticos e Bioequivalncia 1 ASPECTOS QUALITATIVOS 1.1 Absoro de Frmacos 1.2 Distribuio de Frmacos 1.3 Biotransformao de Frmacos 1.4 Eliminao de Frmacos 2 ASPECTOS QUANTITATIVOS 2.1 Modelos Farmacocinticos 2.1.1 Modelos compartimentais 2.1.2 Modelo aberto de um compartimento 2.1.3 Modelos multicompartimentais 2.1.4 Modelo de dois compartimentos 2.1.5 Modelos no compartimentais 3 APLICAES DOS ESTUDOS FARMACOCINTICOS 4 ESTUDOS DE BIOEQUIVALNCIA 4.1 Etapa Clnica 4.2 Etapa Analtica 4.3 Etapa Estatstica e Farmacocintica 5 MEDICAMENTOS GENRICOS CAPTULO 4: Medicamentos Avaliados 1 AMOXICILINA 2 NORFLOXACINO 3 OXCARBAZEPINA CAPTULO 5: Justificativa PARTE II: OBJETIVOS E METODOLOGIA CAPTULO 1: Objetivos 1 OBJETIVO GERAL 2 OBJETIVOS ESPECFICOS CAPTULO 2: Metodologia
52 54 56 57 60 60 61 63 64 65 67 70 70 72 73 73 74 75 78 79 81 82 83 84 84 86 89 91 92 93 93 93 94
1 PROTOCOLO CLNICO 1.1 Local de Confinamento dos Voluntrios 1.2 Delineamento dos Estudos 1.3 Seleo dos voluntrios 1.3.1 Sinais Vitais 1.3.2 Exame cardiolgico 1.3.3 Exames laboratoriais 1.3.4 Valores de referncia dos exames laboratoriais 1.4 Critrios de incluso 1.5 Critrios de excluso 1.6 Critrios para retirada do estudo 1.7 Entrada do voluntrio no estudo 1.8 Restries 1.9 Esquema experimental 1.10 Coleta de Sangue 1.11 Avaliao da segurana 1.12 Aspectos ticos 1.13 Termo de consentimento livre e esclarecido 1.14 Confidencialidade 2 PROTOCOLO EXPERIMENTAL PARA QUANTIFICAO DOS ANALITOS 2.1 Sistema de Qualidade e Biossegurana 2.2 Substncias qumicas e Materiais Utilizados 2.3 Padro Interno 2.4 Preparo das Solues Padro 2.5 Critrios para validao do mtodo bioanaltico 2.5.1 Determinao do limite de quantificao (LQ) 2.5.2 Curva de calibrao/linearidade 2.5.3 Preciso e exatido 2.5.3.1 Preciso e exatido intralote 2.5.3.1 Preciso e exatido interlote 2.5.4 Especificidade 2.5.5 Recuperao
94 94 94 95 95 95 96 97 97 98 99 100 101 101 102 103 104 104 105 105 105 106 107 107 108 108 108 109 110 110 110 110
2.5.6 Estudo de estabilidade do frmaco no fluido biolgico 2.5.6.1 Estabilidade no tempo e condies de anlise (curta durao) 2.5.6.2 Estabilidade do frmaco em ciclos de congelamento e degelo 2.5.6.3 Estabilidade de longa durao 2.6 Desenvolvimento e Validao do Mtodo para Quantificao da Amoxicilina 2.6.1 Equipamentos e condies do HPLC 2.6.2 Preparao de amostras e processo de extrao 2.6.3 Validao do mtodo bioanaltico 2.7 Desenvolvimento e Validao do Mtodo para Quantificao do Norfloxacino 2.7.1 Equipamentos e condies do HPLC 2.7.2 Preparao de amostras e processo de extrao 2.7.3 Validao do mtodo bioanaltico 2.8 Desenvolvimento e Validao do Mtodo para Quantificao da Oxcarbazepina 2.8.1 Equipamentos e condies do HPLC 2.8.2 Preparao de amostras e processo de extrao 2.8.3 Validao do mtodo bioanaltico 2.9 Aplicao dos mtodos propostos em estudos farmacocinticos 2.10 Anlise Estatstica PARTE III: RESULTADOS E DISCUSSO CAPTULO 1: Amoxicilina 1 ETAPA CLNICA 2 DESENVOLVIMENTO E VALIDAO DO MTODO BIOANALTICO PARA QUANTIFICAO DA AMOXICILINA 2.1 Seletividade do mtodo 2.2 Linearidade do mtodo 2.3 Preciso e Exatido 2.4 Recuperao do mtodo 2.5 Estudos de Estabilidade 2 APLICAO DO MTODO BIOANALTICO EM ESTUDO DE
111 112 112 113 114 114 114 115 116 116 116 116 118 118 118 119 120 121 123 124 125 126 126 129 129 130 130 132
BIOEQUIVALNCIA ENTRE DUAS FORMULAES CONTENDO AMOXICILINA (CPSULA DE 500MG) EM VOLUNTRIOS SADIOS CAPTULO 2: Norfloxacino 1 ETAPA CLNICA 2 DESENVOLVIMENTO E VALIDAO DO MTODO 136 136 139 140 140 141 BIOANALTICO PARA QUANTIFICAO DA NORFLOXACINO 2.1 Otimizao dos Procedimentos Gerais 2.2 Linearidade do Mtodo 2.3 Preciso e exatido do mtodo 2.4 Recuperao do mtodo 2.5 Estudos de estabilidade do mtodo 3 APLICAO DO MTODO BIOANALTICO EM ESTUDO DE BIOEQUIVALNCIA ENTRE DUAS FORMULAES CONTENDO NORFLOXACINO (COMPRIMIDO DE 400MG) EM VOLUNTRIOS SADIOS CAPTULO 3: Oxcarbazepina 1 ETAPA CLNICA 2 DESENVOLVIMENTO E VALIDAO DO MTODO 147 147 151 153 154 155 EM ESTUDO 156 158 161 177 183 CONTENDO BIOANALTICO FORMULAO BIOANALTICO PARA QUANTIFICAO DA OXCARBAZEPINA 2.1 Otimizao dos Procedimentos Gerais 2.2 Linearidade do mtodo 2.3 Preciso e exatido do mtodo 2.4 Recuperao do mtodo 2.5 Estudos de estabilidade do mtodo 3 APLICAO DO MTODO DE UMA FARMACOCINTICO 143 144 146 134 135
OXCARBAZEPINA (SUSPENSO 6%) EM VOLUNTRIOS SADIOS CONSIDERAES FINAIS E CONCLUSO REFERNCIAS GLOSSRIO ANEXOS
PARTE I: INTRODUO E REVISO BIBLIOGRFICA
23
PARTE I: INTRODUO E REVISO BIBLIOGRFICA
CAPTULO 1: Validao de Mtodos Bioanalticos
1 INTRODUO
A necessidade de se mostrar a qualidade de medies qumicas, atravs de sua comparabilidade, rastreabilidade e confiabilidade, est sendo cada vez mais reconhecida e exigida. Dados analticos no confiveis podem conduzir a decises desastrosas e a prejuzos financeiros irreparveis. Para garantir que um novo mtodo analtico gere informaes confiveis e interpretveis sobre a amostra, ele deve sofrer uma avaliao denominada validao. A validao de um mtodo um processo contnuo que comea no planejamento da estratgia analtica e continua ao longo de todo o seu desenvolvimento e transferncia. Para registro de novos produtos, todos os rgos reguladores do Brasil e de outros pases exigem a validao de metodologia analtica e, para isso, a maioria deles tem estabelecido documentos oficiais que so diretrizes a serem adotadas no processo de validao (ICH, 1995a; ICH, 1995b; ANVISA, 2003; US-FDA, 2001; US-FDA, 2000; US-FDA, 1994; EURACHEM WORKING GROUP, 1998; THOMPSON et al., 2002; INMETRO, 2003; SHABIR, 2003; ISO/IEC 17025, 1999; USP, 1999; WHO, 1992). Um processo de validao bem definido e documentado oferece s agncias reguladoras evidncias objetivas de que os mtodos e os sistemas so adequados para o uso desejado.
2 CONCEITOS BSICOS DO PROCESSO DE VALIDAO
Vrios autores definem validao de mtodos e pode-se dizer que os conceitos continuam evoluindo e esto constantemente sob considerao pelas agncias reguladoras. Algumas definies podem ser transcritas: - A validao deve garantir, atravs de estudos experimentais, que o mtodo atenda s exigncias das aplicaes analticas, assegurando a confiabilidade dos resultados (ANVISA, 2003).
24
- Validao o processo de definir uma exigncia analtica e confirmar que o mtodo sob investigao tem capacidade de desempenho consistente com o que a aplicao requer (EURACHEM WORKING GROUP, 1998). - Confirmao por testes e apresentao de evidncias objetivas de que determinados requisitos so preenchidos para um dado uso intencional (ISO/IEC 17025, 1999). - A validao de mtodos assegura a credibilidade destes durante o uso rotineiro, sendo algumas vezes mencionado como o processo que fornece uma evidncia documentada de que o mtodo realiza aquilo para o qual indicado para fazer (USP, 1999). - Avaliao sistemtica de um procedimento analtico para demonstrar que est sob as condies nas quais deve ser aplicado (WHO, 1992). Vrios artigos e revises tm sido publicados a respeito de validao de mtodos analticos (THOMPSON et al., 2002; SHABIR, 2003; HUBER, 1998; JENKE, 1997; JENKE, 1998A; JENKE, 1998B; BRUCE et al., 1998; MASSART et al., 1994; SWARTZ; KRULL, 1998; LEITE, 2002; SNYDER et al., 1997), os quais descrevem definies, procedimentos, parmetros e estratgias de validao.
Dentro do mbito geral de validao de mtodos possvel distinguir dois tipos (THOMPSON et al., 2002; MASSART et al., 1994; HILL; REYNOLDS, 1999; VAN DER VOET et al., 1999):
O primeiro, chamado de validao no laboratrio (in house validation), consiste das etapas de validao dentro de um nico laboratrio, seja para validar um mtodo novo que tenha sido desenvolvido localmente ou para verificar que um mtodo adotado de outras fontes est bem aplicado. A validao no laboratrio utilizada nas etapas preliminares do desenvolvimento de uma metodologia e na publicao de artigos para revistas cientficas, em que so avaliadas todas as caractersticas de desempenho da validao da metodologia, porm sem verificar a reprodutibilidade. Pode-se considerar esta etapa como sendo preliminar validao completa (full validation) (THOMPSON et al., 2002).
25
O segundo tipo, validao completa, envolve todas as caractersticas de desempenho e um estudo interlaboratorial que utilizado para verificar como a metodologia se comporta com uma determinada matriz em vrios laboratrios, estabelecendo a reprodutibilidade da metodologia e a incerteza expandida associada metodologia como um todo. S assim a metodologia pode ser aceita como uma metodologia oficial para uma determinada aplicao. Protocolos internacionalmente aceitos tm sido estabelecidos para a validao completa, mais precisamente o Protocolo Harmonizado Internacional (HORWITZ, 1995) e o procedimento ISO (ISO, 1994). Estes protocolos requerem um nmero mnimo de laboratrios e materiais testes para serem includos no estudo interlaboratorial para validao completa do mtodo analtico. No Brasil os estudos de comparaes interlaboratoriais so coordenados pelo Instituto de Pesquisas Tecnolgicas (IPT), atravs do Programa Brasileiro de Metrologia em Qumica.
3 LEGISLAO
Existem razes legais, tcnicas e comerciais que justificam a implantao da validao de mtodos analticos de separao, apesar de no haver uma norma estabelecida de mbito nacional ou internacional. Atualmente, para mostrar competncia tcnica, os laboratrios que executam as anlises devem submeter-se a um credenciamento (accreditation) de um rgo vigente de mbito nacional ou internacional.
No Brasil, h duas agncias credenciadoras para verificar a competncia de laboratrios de ensaios, a ANVISA (Agncia Nacional de Vigilncia Sanitria) e o INMETRO (Instituto Nacional de Metrologia, Normalizao e Qualidade Industrial). Estes rgos disponibilizam guias para o procedimento de validao de mtodos analticos, respectivamente, a Resoluo ANVISA RE n 899, de 29/05/2003 e o guia INMETRO DOQ-CGCRE-008, de maro/2003. Suas similaridades e diferenas podem ser melhor visualizadas no quadro 1.
26
Quadro 1 Parmetros de validao do Inmetro e Anvisa Inmetro
Especificidade/Seletividade Faixa de trabalho e Faixa linear de trabalho Linearidade Limite de Deteco (LD) Limite de Quantificao (LQ) Sensibilidade (inclinao da curva) Exatido e tendncia (bias) Preciso Repetitividade Preciso Intermediria Reprodutibilidade Robustez Incerteza de medio
Anvisa
Especificidade/Seletividade Intervalos da curva de calibrao Linearidade Curva de Calibrao Limite de Deteco (LD) Limite de Quantificao (LQ) Exatido Preciso Repetibilidade (preciso intralote) Preciso intermediria (preciso interlote) Reprodutibilidade (preciso interlaboratorial) Robustez -
importante esclarecer que resolues so documentos com poder de lei, que devem ser obedecidas e guias so documentos que sugerem uma linha a ser seguida e so, portanto, abertos para interpretao. Os guias so recomendaes e so intencionalmente vagos para deixar aos analistas a flexibilidade de adapt-los de acordo com o mtodo a ser usado (KRULL; SWARTZ, 1998).
Os parmetros para validao de mtodos tm sido definidos em diferentes grupos de trabalho de organizaes nacionais ou internacionais. Infelizmente algumas definies so diferentes entre as diversas organizaes. Uma tentativa para harmonizar estas diferenas foi feita para aplicaes farmacuticas, atravs da ICH (ICH, 1995a; ICH, 1995b), na qual representantes das indstrias e agncias reguladoras dos EUA, Europa e Japo definiram parmetros,
27
requerimentos e, em alguns casos, tambm metodologias para validao dos mtodos analticos.
A IUPAC (International Union of Pure and Applied Chemistry) tambm redigiu um documento tcnico que define um guia para validao de mtodos analticos que tem sido utilizado pela ISO10. A norma internacional ISO/IEC 17025, que uma norma especfica para laboratrios de ensaio e de calibrao, apresenta a validao de mtodos como um dos requisitos tcnicos importantes na qualidade assegurada dos laboratrios de ensaio, bem como a documentao do trabalho de validao (ISO, 1999). O US-FDA (United States Food and Drug Administration) tambm tem proposto guias sobre validao de mtodos (FDA, 1993).
Assim, rgos como ICH, IUPAC, ISO, ANVISA, INMETRO e outros exigem o item validao de mtodos analticos como um requisito fundamental no credenciamento para qualidade assegurada e demonstrao de competncia tcnica (ICH, 1995a; ICH, 1995b; ANVISA, 2003; FDA, 1985; FDA, 1998a, FDA, 1998b; THOMPSON et al., 2002; INMETRO, 2003). O que se pode observar que no h um procedimento normatizado que estabelea como executar a validao de mtodos instrumentais de separao. Como estes organismos so responsveis por acompanhar e credenciar a competncia de laboratrios de ensaios, importante ressaltar que as diferentes terminologias e at algumas caractersticas de desempenho do mtodo tm, em sua maior parte, o mesmo significado, porm descrito de uma maneira distinta, para aplicaes diferentes.
4 PROCESSO DE VALIDAO
essencial que os estudos de validao sejam representativos e conduzidos de modo que a variao da faixa de concentrao e os tipos de amostras sejam adequados. Um mtodo para um composto majoritrio requer um critrio de aceitao e uma abordagem diferente de um mtodo desenvolvido para anlise de traos. A freqncia com que o mtodo ser utilizado (muitas vezes em um dia, uma vez em um dia para um estudo rpido, uma vez em um ms, etc.) tambm influencia o tipo de estudo de validao que necessrio. Os parmetros
28
analticos devem ser baseados na inteno do uso do mtodo. Por exemplo, se um mtodo ser usado para anlise qualitativa em nvel de traos, no h necessidade de testar e validar a linearidade do mtodo sobre toda a faixa linear dinmica do equipamento. O objetivo do mtodo pode incluir tambm os diferentes tipos de equipamentos e os lugares em que o mtodo ser utilizado, ou seja, se o mtodo desenvolvido para ser utilizado em instrumento e laboratrio especficos, no h necessidade de usar instrumentos de outras marcas ou incluir outros laboratrios nos experimentos de validao. Desta forma, os experimentos podem ser limitados para o que realmente necessrio (RIBANI et al., 2004).
5 PARMETROS ANALTICOS PARA VALIDAO DE MTODOS
Os parmetros analticos normalmente encontrados para validao de mtodos de separao so: seletividade; linearidade e faixa de aplicao; preciso; exatido; limite de deteco; limite de quantificao e robustez. Estes termos so conhecidos como parmetros de desempenho analtico (SWARTZ; KRULL, 1998), caractersticas de desempenho (THOMPSON et al., 2002; INMETRO, 2003) e, algumas vezes, como figuras analticas de mrito (SWARTZ; KRULL, 1998).
5.1 Seletividade
A seletividade de um mtodo instrumental de separao a capacidade de avaliar, de forma inequvoca, as substncias em exame na presena de componentes que podem interferir com a sua determinao em uma amostra complexa. A seletividade avalia o grau de interferncia de espcies como outro ingrediente ativo, excipientes, impurezas e produtos de degradao, bem como outros compostos de propriedades similares que possam estar, porventura, presentes. A seletividade garante que o pico de resposta seja exclusivamente do composto de interesse (USP, 1999; VESSMAN et al., 2001). Se a seletividade no for assegurada, a linearidade, a exatido e a preciso estaro seriamente comprometidas.
29
O mesmo significado tem sido freqentemente utilizado para o termo especificidade (ICH, 1995; SHABIR, 2003; USP, 1999; BRUCE et al., 1998; MASSART, et al., 1994). Esta situao gera confuso desnecessria e isto pode ser evitado utilizando somente o termo seletividade, como sugerido pela IUPAC (VESSMAN et al., 2001). Um mtodo instrumental de separao que produz resposta para uma nica substncia de interesse, normalmente um dado elemento, pode ser chamado de especfico e um mtodo que produz resposta para vrios compostos qumicos, com uma caracterstica em comum, pode ser chamado de seletivo (HUBER, 1998; BRUCE et al., 1998). Desde que h poucos mtodos cromatogrficos que respondem a apenas uma substncia, o termo seletividade mais apropriado, como sugerido pela IUPAC.
A seletividade o primeiro passo no desenvolvimento e validao de um mtodo instrumental de separao e deve ser reavaliada continuamente durante a validao e subseqente uso do mtodo. Algumas amostras podem sofrer degradao, gerando compostos que no foram observados inicialmente, que podem coeluir com a substncia de interesse.
A seletividade pode ser obtida de vrias maneiras. A primeira forma de se avaliar a seletividade comparando a matriz isenta da substncia de interesse e a matriz adicionada com esta substncia (padro), sendo que, nesse caso, nenhum interferente deve eluir no tempo de reteno da substncia de interesse, que deve estar bem separada dos demais compostos presentes na amostra (CODEX, 1995; ICH, 1995; SHABIR, 2003; SWARTZ; KRULL, 1998). Uma segunda maneira atravs da avaliao com detectores modernos (arranjo de diodos, espectrmetro de massas), que comparam o espectro do pico obtido na separao com o de um padro e utiliza-se isto como uma indicao da presena do composto puro (HUBER, 1998; JENKER, 1998a; VESSMAN et al., 2001). Estas duas maneiras so as mais utilizadas. O mtodo de adio padro tambm pode ser aplicado para os estudos de seletividade (USP, 1999; JENKER, 1998b), porm este mtodo utilizado quando no possvel obter a matriz isenta da substncia de interesse. Neste caso feita uma curva analtica com adio da substncia de interesse na amostra e comparada com uma curva analtica sem a presena da matriz.
30
Comparam-se ento as duas curvas analticas e caso elas sejam paralelas, pode-se dizer que no h interferncia da matriz na determinao da substncia de interesse, portanto o mtodo seletivo. Outro procedimento para avaliar a seletividade atravs da coleta do composto de interesse e realizao de nova anlise por outra tcnica cromatogrfica, ou com mtodos e tcnicas que so especficos para a estrutura da substncia de interesse como, por exemplo, espectrometria de massas, ressonncia magntica nuclear, espectroscopia no infravermelho ou bioensaios especficos (JENKER, 1998a).
5.2 Linearidade e Faixa de Aplicao
A linearidade corresponde capacidade do mtodo em fornecer resultados diretamente proporcionais concentrao da substncia em exame, dentro de uma determinada faixa de aplicao (ICH, 1995b; USP, 1999; SWARTZ; KRULL, 1998).
A correlao entre o sinal medido (rea ou altura do pico) e a massa ou concentrao da espcie a ser quantificada muito raramente conhecida a priori. Na maior parte dos casos, a relao matemtica entre o sinal e a concentrao ou massa da espcie de interesse deve ser determinada empiricamente, a partir de sinais medidos para massas ou concentraes conhecidas dessa espcie (AUGUSTO et al., 2000). Essa relao matemtica, muitas vezes, pode ser expressa como uma equao de reta chamada de curva analtica (BARROS NETO et al., 2002). Um exemplo de curva analtica pode ser visto na Figura 1a. Embora somente dois pontos definam uma reta, na prtica as linhas devem ser definidas por no mnimo cinco pontos que no incluam o ponto zero na curva, devido aos possveis erros associados (THOMPSON et al., 2002).
Matematicamente, a estimativa dos coeficientes de uma curva analtica a partir de um conjunto de medies experimentais pode ser efetuada usando o mtodo matemtico conhecido como regresso linear (CUSTODIO et al., 1997). Alm dos coeficientes de regresso a e b, tambm possvel calcular, a partir dos pontos experimentais, o coeficiente de correlao r. Este parmetro permite uma
31
estimativa da qualidade da curva obtida, pois quanto mais prximo de 1,0, menor a disperso do conjunto de pontos experimentais e menor a incerteza dos coeficientes de regresso estimados. Para verificar se a equao de regresso estatisticamente significativa podem ser efetuados os testes de ajuste do modelo linear, validade da regresso, sua eficincia e sua eficincia mxima (CHUI et al., 2001; BARROS NETO et al., 2001). Um coeficiente de correlao maior que 0,999 considerado como evidncia de um ajuste ideal dos dados para a linha de regresso (SHABIR, 2003; JENKER, 1998a; GREEN, 1996). A ANVISA (2003) recomenda um coeficiente de correlao igual a 0,99 e o INMETRO (2003) um valor acima de 0,90.
Em qualquer tcnica instrumental, a relao linear simples, descrita pela equao y = ax + b, s vlida em um determinado intervalo de massa ou concentrao da espcie medida. Este intervalo de massas ou concentraes, no qual se pode construir uma curva analtica linear, a faixa linear dinmica (AUGUSTO et al., 2000). Ainda que as causas para a perda de linearidade sejam caractersticas de cada tcnica, este um fenmeno que pode ocorrer com qualquer conjunto de dados. Assim, o clculo dos coeficientes de regresso de uma curva analtica deve ser acompanhado de uma cuidadosa inspeo, para verificar se todos os pontos a serem usados esto dentro da faixa linear dinmica correspondente. Augusto et al. (2000) descreveram uma forma de calcular se os pontos de uma curva analtica esto inseridos na faixa linear, baseando-se em relaes geomtricas do grfico de regresso. Uma terceira abordagem dividir os dados do sinal pelas suas respectivas concentraes, fornecendo as respostas relativas (HUBER, 1998). Um grfico construdo com as respostas relativas no eixo y e as concentraes correspondentes em escala logartmica no eixo x. A linha obtida deve ser horizontal sobre toda a faixa linear. So desenhadas outras linhas horizontais paralelas no grfico, para 95 e 105% da linha da faixa linear. Conclui-se que o mtodo linear at o ponto onde a resposta relativa intercepta a linha de 95 ou 105%. A Figura 1 mostra uma comparao da determinao do intervalo linear dinmico atravs da curva analtica clssica (Figura 1a) e da sua representao logartmica (Figura 1b). A construo da curva com a concentrao em escala logartmica permite melhor visualizao da faixa linear.
32
Figura 1. Determinao grfica das curvas de linearidade atravs da: (a) curva analtica clssica; (b) grfico da razo sinal/concentrao vs. concentrao em escala logartmica. A faixa de aplicao corresponde ao intervalo entre o valor superior e inferior da substncia em exame, que atenda aos requisitos de preciso e exatido (SWARTZ; KRULL, 1998). A faixa de aplicao normalmente expressa nas mesmas unidades dos resultados obtidos pelo mtodo, e depende do uso em questo. Vrias recomendaes so encontradas na literatura. Por exemplo, a ANVISA especifica um intervalo compreendido entre 80-120% da concentrao terica para frmacos e medicamentos e de at 120% do limite mximo especificado para determinao de impurezas (ANVISA, 2003). Para resduos, o GARP (ASSOCIAO GRUPO DE ANALISTAS DE RESDUOS DE PESTICIDAS, 1999) recomenda uma faixa de concentrao com valores variando entre a metade e o quntuplo da concentrao do limite de quantificao. A IUPAC especifica que os pontos da curva analtica devem ser igualmente espaados sobre a faixa de concentrao de interesse e que esta faixa compreenda 0150% ou 50150% do valor esperado, dependendo de qual destas duas opes for mais adequada (THOMPSON et al., 2002). Para produtos formulados, a ICH, entre outros, recomenda uma variao de 20% do valor declarado ou esperado (ICH, 1995b; USP, 1994; SHABIR, 2003).
As diretrizes da ICH (1995b) e da ANVISA (2003) especificam um mnimo de cinco nveis de concentrao, juntamente com certos mnimos de variao especificados. O GARP tambm sugere cinco concentraes que devem ser
33
injetadas no equipamento em ordem crescente de concentrao, no mnimo trs vezes cada, com estimativa do desvio padro relativo (RSD) entre as injees inferior a 5%. A IUPAC recomenda seis ou mais nveis de concentrao (THOMPSON et al., 2002).
A quantificao do composto de interesse em validao pode ser obtida atravs dos seguintes mtodos: padronizao externa; padronizao interna; superposio de matriz; adio padro.
5.2.1 Padronizao externa
O mtodo de padronizao externa compara a rea da substncia a ser quantificada na amostra com as reas obtidas com solues de concentraes conhecidas preparadas a partir de um padro. Preparam-se solues da substncia a ser quantificada em diversas concentraes; obtm-se o cromatograma correspondente a cada uma delas e, em um grfico, relacionam-se as reas obtidas com as concentraes. Utilizando este grfico ou a equao da curva resultante, pode-se calcular a concentrao desta substncia na amostra a partir da rea da substncia obtida no cromatograma resultante de uma injeo separada. Este mtodo sensvel a erros de preparo das amostras e dos padres e de injeo das solues padro e das amostras (SWARTZ; KRULL, 1998; CUADROS-RODRGUEZ et al., 2001) e por isso deve ser feito a cada anlise.
5.2.2 Padronizao interna
O mtodo de padronizao interna consiste na preparao das solues padro de concentraes conhecidas da substncia de interesse, s quais se adiciona a mesma quantidade conhecida de um composto chamado padro interno. Aps anlise dessas solues, constri-se um grfico, relacionando a razo de reas (rea da substncia/rea do padro interno que tem concentrao constante) com a concentrao (variada) da substncia. A amostra tambm analisada aps a adio da mesma quantidade conhecida do padro interno. Atravs da razo de reas obtidas no cromatograma tem-se a concentrao da substncia na amostra.
34
Idealmente, a substncia usada como padro interno deve ser similar substncia a ser quantificada, ter tempo de reteno prximo a esta substncia, no reagir com a substncia ou outro componente da matriz, no fazer parte da amostra e, quando cromatografada, ficar separada de todas as demais substncias presentes na amostra (SWARTZ; KRULL, 1998; CUADROS-RODRGUEZ et al., 2001). Este ltimo requisito no necessrio quando a deteco feita por espectrometria de massas, na qual cada composto produz um espectro caracterstico. O mtodo de padronizao interna extremamente til, especialmente pelo fato de que independe de pequenas mudanas em variveis experimentais, como temperatura da coluna e tamanho da amostra. Este mtodo bastante til em cromatografia gasosa, na qual se usa seringa para injeo de amostra (LANAS, 1993) e por isso deve ser feito a cada anlise.
5.2.3 Superposio de matriz
O mtodo de superposio de matriz (matrix-matched) consiste na adio do padro da substncia em diversas concentraes em uma matriz similar da amostra, isenta da substncia, e construo do grfico de calibrao relacionando as reas obtidas com as concentraes dos padres. O mtodo de superposio de matriz pode ser utilizado para calibrao, tanto com a padronizao interna como com a padronizao externa. Este mtodo usado para compensar o efeito da matriz ou de possveis interferentes e de suma importncia em determinaes quando a matriz pode interferir na pr-concentrao, extrao, separao ou deteco da substncia de interesse. Sua principal vantagem sobre o mtodo de padronizao externa que fornece uma melhor correspondncia com a composio da amostra. Por exemplo, se algumas substncias so determinadas em soro humano e uma soluo padro aquosa for usada na calibrao, resultados errneos podem ser obtidos por causa do efeito da matriz; para tais medies, uma matriz de soro humano seria melhor para realizar a calibrao do que a soluo aquosa (CUADROS-RODRGUEZ et al., 2001). O mtodo de superposio de matriz tem o inconveniente de no proporcionar a magnitude do efeito de co-extratos, alm de aumentar o custo e o tempo das anlises (EGEA-GONZLEZ et al., 2002). Alguns autores acreditam que o efeito dos coextratos sobre a resposta da
35
substncia de interesse deveria ser avaliado pela comparao do mtodo de superposio de matriz com a padronizao externa (padres preparados nos solventes) (HILL; REYNOLDS, 1999). Apesar de se obter uma calibrao confivel com o mtodo de superposio da matriz, ele somente uma forma para compensar efeitos da matriz, mas no elimina situaes analticas tpicas: a intensidade de um efeito e a concentrao de interferentes na matriz podem diferir de uma matriz ou amostra para outra (CUADROS-RODRGUEZ et al., 2003). Assim, em amostras nas quais pode ocorrer o efeito da matriz e no se tem disponvel uma matriz isenta da substncia de interesse para utilizar o mtodo de superposio de matriz, deve-se utilizar o mtodo de adio padro (CUADROS-RODRGUEZ et al., 2003).
5.2.4 Adio padro
O mtodo de adio padro consiste na adio de quantidades conhecidas da substncia de interesse que est sendo analisada a quantidades conhecidas da amostra, antes do seu preparo. Estas amostras com o padro incorporado so utilizadas para a obteno dos cromatogramas. Constri-se uma curva analtica relacionando as quantidades da substncia adicionada amostra com as respectivas reas obtidas. O ponto onde a reta corta o eixo das ordenadas corresponde rea do pico da substncia que est sendo determinada, sem qualquer adio do padro. A extrapolao da reta define, no eixo das abscissas, a concentrao da substncia na amostra analisada (BERG et al., 1988). O mtodo de adio padro trabalhoso, mas especialmente importante quando a amostra muito complexa, quando as interaes com a matriz so significativas e quando houver dificuldade de encontrar um padro interno adequado ou uma matriz isenta da substncia de interesse (SNYDER et al., 1997).
Os mtodos de quantificao no tm regras ou guias, exceto que o mtodo final selecionado deve fornecer a melhor exatido possvel e um alto nvel de preciso. O mtodo escolhido para quantificao deve atingir estes objetivos em um menor tempo possvel, com um mnimo de envolvimento do operador, alm de utilizar pouca quantidade de amostra. O mtodo de quantificao ideal depender da
36
amostra especfica, do nmero de amostras, da complexidade da matriz, da possibilidade de automao e da disponibilidade de padres.
Quanto aos aspectos prticos, o preparo das solues padro pode ser realizado de diversas maneiras. Em uma delas, prepara-se uma soluo mais concentrada, a partir da pesagem do padro, denominada soluo estoque, que deve ser preparada de acordo com o tempo de estabilidade da substncia em soluo ou pelo menos a cada seis meses. As solues preparadas por diluio so chamadas solues de trabalho e recomenda-se que sejam preparadas pelo menos a cada duas ou trs semanas (GARP, 1999). Duas abordagens ocorrem no preparo das solues por diluio. Na primeira, as solues de trabalho podem partir de uma nica soluo estoque, atravs de diluies sucessivas das solues de trabalho anteriormente preparadas ou atravs do preparo de todas as solues diludas, partindo sempre da soluo estoque. Este ltimo modo o mais recomendado, porm, dependendo da faixa de concentrao em questo, diluies diretamente da soluo estoque podem envolver medies de volumes to pequeno que o erro se torna grande. A Figura 2a mostra um esquema da seqncia utilizando este modo de preparo. Outra abordagem para o preparo das solues de trabalho mostrada na Figura 2b. Neste caso, so preparadas duas solues estoque de concentraes diferentes e as solues de trabalho so preparadas a partir destas duas solues, seja por diluies sucessivas ou partindo das solues estoque. Este modo de preparo das solues de trabalho mais adequado, pois permite avaliar atravs da curva analtica o erro embutido na soluo estoque, decorrente da pesagem dos padres.
37
Figura 2. Modos de diluio da soluo estoque: a) diluio a partir de uma soluo estoque e b) diluio a partir de duas solues estoques (1 e 2) preparadas com concentraes diferentes. Outra maneira de se preparar as solues seria atravs da pesagem separada de cada concentrao do padro da substncia da curva analtica, ao invs da utilizao de diluies sucessivas de uma soluo mais concentrada (SWARTZ; KRULL, 1998). Esta a maneira ideal de se preparar as solues, pois a preparao individual mostra o erro verdadeiro na preparao de todas as solues padro e no, erros de diluio. Entretanto, este mtodo torna-se invivel quando se trabalha com anlise de traos, j que a quantidade de padro a ser pesada to pequena que a sensibilidade da balana no permite tais pesagens, devido aos erros associados.
5.3 Preciso
Representa a disperso de resultados entre ensaios independentes, repetidos de uma mesma amostra, amostras semelhantes ou padres, sob condies definidas (ICH, 1995b; INMETRO, 2003). A preciso avaliada pelo desvio padro absoluto (), que utiliza um nmero significativo de medies, normalmente maior que 20. Na prtica, em validao de mtodos, o nmero de determinaes geralmente pequeno e o que se calcula a estimativa do desvio padro absoluto (s).
38
Outra expresso da preciso atravs da estimativa do desvio padro relativo (RSD), tambm conhecido como coeficiente de variao (CV).
RSD (%) ou CV (%) = s/x * 100, onde x a mdia aritmtica de um pequeno nmero de medies (mdia das determinaes).
Normalmente,
mtodos
que
quantificam
compostos
em
macro
quantidades requerem um RSD de 1 a 2%. Em mtodos de anlise de traos ou impurezas, so aceitos RSD de at 20%, dependendo da complexidade da amostra (HUBER, 1998). Uma maneira simples de melhorar a preciso aumentar o nmero de replicatas.
A preciso em validao de mtodos considerada em trs nveis diferentes: repetitividade; preciso intermediria; reprodutibilidade.
5.3.1 Repetitividade
A repetitividade (repeatability) representa a concordncia entre os resultados de medies sucessivas de um mesmo mtodo, efetuadas sob as mesmas condies de medio, chamadas condies de repetitividade: mesmo procedimento; mesmo analista; mesmo instrumento usado sob as mesmas condies; mesmo local; repeties em um curto intervalo de tempo. O termo repetitividade adotado pelo Vocabulrio Internacional de Metrologia (INMETRO, 2000), sendo utilizado pelo INMETRO. Por outro lado, a ANVISA utiliza o mesmo conceito para o termo repetibilidade (ANVISA, 2003).
A repetitividade envolve vrias medies da mesma amostra, em diferentes preparaes e , algumas vezes, denominada preciso intra-ensaio (SHABIR, 2003; GREEN, 1996) ou intralote. Pode ser expresso atravs da estimativa do desvio padro relativo (RSD).
No se deve confundir repetitividade com preciso instrumental, a qual medida pelas injees repetitivas, seqenciais da mesma amostra (tipicamente 10
39
ou mais vezes), seguida pela mdia dos valores da rea do pico ou altura do pico e determinao da estimativa do desvio padro relativo de todas as injees.
Para a repetitividade, o INMETRO (2003) recomenda sete ou mais repeties para o clculo da estimativa do desvio padro. A ICH (1995b) e ANVISA (2003) sugerem que a repetitividade seja verificada a partir de um mnimo de nove determinaes cobrindo o limite especificado do procedimento (ex.: trs nveis, trs repeties cada um), ou a partir de um mnimo de seis determinaes a uma concentrao similar ao valor esperado.
5.3.2 Preciso intermediria
Indica o efeito das variaes dentro do laboratrio devido a eventos como diferentes dias ou diferentes analistas ou diferentes equipamentos ou uma combinao destes fatores (ICH, 1995b).
A preciso intermediria reconhecida como a mais representativa da variabilidade dos resultados em um nico laboratrio e, como tal, mais aconselhvel de ser adotada. O objetivo da validao da preciso intermediria verificar que no mesmo laboratrio o mtodo fornecer os mesmos resultados.
O nmero de ensaios necessrios para se avaliar a preciso intermediria segue a mesma recomendao da ICH (1995b) e ANVISA (2003) para o clculo de repetitividade descrita acima. A preciso intermediria pode ser expressa atravs da estimativa do desvio padro relativo (RSD).
5.3.3 Reprodutibilidade
o grau de concordncia entre os resultados das medies de uma mesma amostra, efetuadas sob condies variadas (mudana de operador, local, equipamentos, etc.) (INMETRO, 2000). A reprodutibilidade refere-se aos resultados dos estudos de colaborao entre laboratrios e deve ser considerada em situaes como a padronizao de procedimentos analticos a serem includos, por exemplo,
40
em farmacopias, procedimentos do CODEX, etc. muito comum encontrar desacordo entre mtodos analticos. Isto aparece quando vrios laboratrios analisam uma amostra em comum, em estudos colaborativos. Freqentemente, altas variaes so observadas entre os resultados. Assim, os dados provenientes de apenas um laboratrio no so suficientes para avaliar a reprodutibilidade do mtodo. Estudos colaborativos no so somente indispensveis para avaliao da reprodutibilidade, eles tambm podem ser de grande ajuda para testar a exatido do mtodo (VIAL; JARDY, 2001).
A IUPAC no aconselha tirar concluses com menos de cinco laboratrios e recomenda oito laboratrios em seu guia atual (THOMPSON et al., 2002). Alm disso, mais crtico que o nmero de laboratrios envolvidos que estes possuam competncia e habilidades similares queles que usaro o mtodo em rotina.
A documentao que apia os estudos de preciso em nvel de reprodutibilidade deve incluir estimativa do desvio padro absoluto, estimativa do desvio padro relativo e intervalo de confiana. Horwitz et al. (1980) estabeleceram uma relao matemtica para expressar a dependncia entre valores do RSD e concentrao da substncia, pelo exame de resultados cumulativos de estudos colaborativos envolvendo grande faixa de compostos de interesse, matrizes e tcnicas analticas. Os valores obtidos por esta relao matemtica so introduzidos em um grfico e originam a denominada Trombeta de Horwitz (HORWITZ et al., 1980).
5.4 Exatido
Representa o grau de concordncia entre os resultados individuais encontrados em um determinado ensaio e um valor de referncia aceito como verdadeiro (ICH, 1995b; INMETRO, 2003). importante observar que um valor exato ou verdadeiro o valor obtido por uma medio perfeita e este valor indeterminado por natureza (ISO, 1993).
41
A exatido sempre considerada dentro de certos limites, a um dado nvel de confiana (ou seja, aparece sempre associada a valores de preciso). Estes limites podem ser estreitos em nveis de concentrao elevados e mais amplos em nveis de traos.
O nmero de ensaios varia segundo a legislao ou diretriz adotada e tambm com as caractersticas da pesquisa. A ICH (1995b) estabelece que um mnimo de nove determinaes envolvendo um mnimo de trs diferentes nveis de concentrao deve ser obedecido. Por exemplo, ensaios em triplicata para trs nveis de concentrao. Esta recomendao tambm adotada pela ANVISA (2003).
Os processos mais utilizados para avaliar a exatido de um mtodo so: materiais de referncia; comparao de mtodos; ensaios de recuperao; adio padro.
5.4.1 Materiais de referncia certificados (CRM)
Os CRM so materiais de referncia acompanhados de um certificado que possui o valor de concentrao de uma dada substncia, ou outra grandeza para cada parmetro e uma incerteza associada. Os materiais de referncia certificados so fornecidos por organismos reconhecidos e confiveis, como NIST (National Institute of Standards and Technology - USA), LGC (Laboratory of the Government Chemist - UK), USP, FAPAS (Food Analysis Performance Assessment Scheme - UK), etc.
Os valores obtidos pelo laboratrio (a mdia e a estimativa do desvio padro de uma srie de replicatas) da mesma amostra padro devem ser comparados com os valores certificados do material de referncia, para verificar a exatido do mtodo.
42
5.4.2 Comparao de mtodos
Consistem na comparao entre resultados obtidos empregando-se o mtodo em desenvolvimento e os resultados conseguidos atravs de um mtodo de referncia, avaliando o grau de proximidade entre os resultados obtidos pelos dois mtodos, ou seja, o grau de exatido do mtodo testado em relao ao de referncia. Esta abordagem assume que a incerteza do mtodo de referncia conhecida.
As anlises so efetuadas em replicata, utilizando os dois mtodos em separado (o mtodo em desenvolvimento e o mtodo de referncia), sobre as mesmas amostras, em uma faixa de concentraes em que se pretende validar o mtodo.
5.4.3 Ensaios de recuperao
A recuperao (ou fator de recuperao), R, definida como a proporo da quantidade da substncia de interesse, presente ou adicionada na poro analtica do material teste, que extrada e passvel de ser quantificada (THOMPSON et al., 1999). A informao de recuperao pode ser estimada de CRM (em que a quantidade de substncia previamente conhecida), quando disponveis, ou de um composto substituto (surrogate). O substituto definido como um composto ou elemento puro adicionado ao material teste, no qual o comportamento qumico e fsico representativo da substncia de interesse na forma nativa (CUADROS-RODRGUEZ et al., 2001). Diz-se que o composto um substituto porque este transferido para a amostra e pode no estar efetivamente no mesmo equilbrio que se encontra a substncia na forma nativa, ento se determina a recuperao do substituto, fazendo uma correo de recuperao para a substncia de interesse (THOMPSON et al., 1999). Os compostos substitutos, adicionados nas amostras, podem ser de vrios tipos: padro da substncia adicionado matriz isenta da substncia ou amostra (fortificao, incorporao, dopagem, enriquecimento, termos provenientes
43
do ingls spiking); o US-FDA reconhece duas categorias de padres de referncia: compendiais e no compendiais. Os padres de referncia compendiais so obtidos de fontes como a USP e no necessitam de caracterizao posterior. Os padres de referncia no compendiais so substncias com elevado teor de pureza, que podem ser obtidas atravs de um esforo razovel e devem ser cuidadosamente caracterizados para garantir sua identidade, potncia e pureza. recomendvel que fatores de correo de pureza sejam includos em qualquer clculo existente no mtodo. uma verso da substncia modificada isotopicamente e composto quimicamente diferente da substncia de interesse, mas representativo de seu comportamento. Algumas vezes este composto denominado padro interno (LEITE, 2002; CUADROS-RODRGUEZ et al., 2001). A limitao do procedimento de recuperao a de que a substncia adicionada no est, necessariamente, na mesma forma que a presente na amostra. Isso pode implicar, por exemplo, na presena de substncias adicionadas em uma forma que proporcione melhor deteco, ocasionando avaliaes excessivamente otimistas da recuperao. Pelo fato de outros componentes da matriz poderem interferir na separao, deteco ou na quantificao da substncia, efeitos dos componentes da matriz devem ser investigados.
importante considerar como a eficincia do mtodo varia em funo da concentrao da substncia. Na maioria dos casos, a disperso dos resultados aumenta com a diminuio da concentrao e a recuperao pode diferir substancialmente a altas e baixas concentraes.
Por esse motivo, a recuperao deve ser avaliada na faixa de concentrao esperada para o composto de interesse. Isto pode ser feito adicionando a substncia em pelo menos trs diferentes concentraes, por exemplo, prximo ao limite de quantificao, prximo concentrao mxima permitida pelo mtodo em teste e em uma concentrao prxima mdia da faixa de uso do mtodo. Para anlises em nvel de resduos, o GARP recomenda que se trabalhe nos nveis de adio de 1, 2 e 10 vezes o valor de limite de quantificao (GARP, 1999). Para componentes em maiores concentraes, os nveis de adio
44
podem ser 50, 75, 100, 125 e 150% do nvel esperado para a substncia (SNYDER et al., 1997).
As medies de recuperao so as mais comuns devido dificuldade em se obterem CRM e so expressas em termos de porcentagem da quantidade medida da substncia em relao quantidade adicionada na matriz (branco ou placebo), em um determinado nmero de ensaios (BURNS et al., 2002).
Os intervalos aceitveis de recuperao para anlise de resduos geralmente esto entre 70 e 120%, com preciso de at 20%. Porm, dependendo da complexidade analtica e da amostra, este valor pode ser de 50 a 120%, com preciso de at 15% (GARD, 1999).
5.4.4 Adio padro
Este mtodo usado quando for difcil ou impossvel preparar um branco da matriz sem a substncia de interesse. Um exemplo seria a anlise de -caroteno em amostras de mamo, nas quais o -caroteno sempre estar presente.
No mtodo de adio padro, quantidades conhecidas da substncia so adicionadas em diferentes nveis numa matriz da amostra, antes do procedimento de preparo da amostra, que j contenha quantidades (desconhecidas) da substncia (DE ANDRADE, 1987). A concentrao da substncia de interesse na amostra original pode ser determinada grfica e matematicamente, como j mostrado anteriormente. Em geral, para adio padro, uma boa abordagem adicionar 25, 50 e 100% da concentrao esperada da substncia na matriz (SNYDER et al., 1997). A amostra sem adio do padro e cada uma das amostras com o padro adicionado devem ser analisadas e as quantidades medidas relacionadas com a quantidade adicionada.
45
5.5 Limite de Deteco (LD)
O limite de deteco (LD) representa a menor concentrao da substncia em exame que pode ser detectada, mas no necessariamente quantificada, utilizando um determinado procedimento experimental (ICH, 1995b; INMETRO, 2003).
O LD pode ser calculado de trs maneiras diferentes: mtodo visual, mtodo relao sinal-rudo, mtodo baseado em parmetros da curva analtica.
5.5.1 Mtodo visual
utilizado para determinar o limite de deteco utilizando a matriz com adio de concentraes conhecidas da substncia de interesse, de tal modo que se possa distinguir entre rudo e sinal analtico pela visualizao da menor concentrao visvel (detectvel). Este procedimento tambm pode ser feito atravs do instrumento utilizando parmetros de deteco no mtodo de integrao.
5.5.2 Mtodo da relao sinal-rudo
Este mtodo pode ser aplicado somente em procedimentos analticos que mostram o rudo da linha de base. Para determinar a relao sinal-rudo, feita a comparao entre a medio dos sinais de amostras em baixas concentraes conhecidas do composto de interesse na matriz e um branco (matriz isenta do composto de interesse) destas amostras. Assim, estabelecida uma concentrao mnima na qual a substncia pode ser facilmente detectada. A relao sinal-rudo pode ser de 3:1 ou 2:1, propores geralmente aceitas como estimativas do limite de deteco.
46
5.5.3 Mtodo baseado em parmetros da curva analtica
O limite de deteco (LD) pode ser expresso como:
LD = 3,3 x s/S
onde s a estimativa do desvio padro da resposta, que pode ser a estimativa do desvio padro do branco, da equao da linha de regresso ou do coeficiente linear da equao e S a inclinao (slope) ou coeficiente angular da curva analtica.
Para calcular estes dados, uma curva analtica dever ser feita utilizando a matriz contendo o composto de interesse na faixa de concentrao prxima ao limite de deteco. Softwares como Microsoft Excel ou Microcal Origin podem calcular os parmetros da curva e a estimativa do desvio padro relativo destes parmetros. Para adquirir melhor compreenso dos clculos envolvidos, podem-se consultar livros de estatstica (BARROS NETO et al., 2002; MILLER; MILLER, 1998; BOX et al., 1987).
5.6 Limite de Quantificao (LQ)
O limite de quantificao (LQ) representa a menor concentrao da substncia em exame que pode ser medida, utilizando um determinado procedimento experimental (ICH, 1995b; INMETRO, 2003).
Como o LD, o LQ expresso como uma concentrao, sendo que a preciso e exatido das determinaes tambm devem ser registradas. Esse critrio uma boa regra a ser seguida, porm no se deve esquecer que a determinao do LQ representa um compromisso entre a concentrao, a preciso e a exatido exigidas. Isto significa que, quando decresce o nvel de concentrao do LQ, a medio torna-se menos precisa. Se houver necessidade de maior preciso, uma concentrao maior deve ser registrada para o LQ. O mtodo analtico e seu respectivo uso ditam esse compromisso.
47
Os mesmos critrios de LD podem ser adotados para o LQ, utilizando a relao 10:1, ou seja, o LQ pode ser calculado utilizando o mtodo visual, a relao sinal-rudo ou a relao entre a estimativa do desvio padro da resposta (s) (que pode ser a estimativa do desvio padro do branco, da equao da linha de regresso ou do coeficiente linear da equao) e a inclinao da curva analtica (S), em nveis prximos ao LQ, a partir da equao:
LQ = 10 x s/S
O mtodo mais utilizado o da relao sinal-rudo para tcnicas analticas em geral, porm em tcnicas analticas de separao, como as cromatogrficas e eletroforticas, a medio do rudo no trivial e s vezes subjetiva (j que a curva analtica construda com a rea e no somente o sinal do detector). Alm disso, tanto o LD quanto o LQ podem ser afetados pelas condies cromatogrficas. Picos maiores aumentam a relao sinal-rudo, resultando em LD e LQ mais baixos. Alm disso, a determinao cromatogrfica desses parmetros deve considerar tanto o tipo quanto o tempo de uso da coluna.
O melhor caminho para resolver este problema do clculo do LD e LQ utilizar o mtodo baseado nos parmetros da curva analtica, que estatisticamente mais confivel. A curva analtica deve conter a concentrao correspondente ao LQ.
5.7 Robustez
De acordo com o INMETRO (2003), a robustez de um mtodo (robustness) mede a sensibilidade que este apresenta em face de pequenas variaes. Diz-se que um mtodo robusto quando ele no afetado por uma modificao pequena e deliberada em seus parmetros. A robustez de um mtodo cromatogrfico avaliada, por exemplo, pela variao de parmetros como a concentrao do solvente orgnico, pH e fora inica da fase mvel em HPLC, programao da temperatura, natureza do gs de arraste em GC, bem como o tempo de extrao, agitao, etc. As mudanas introduzidas refletem as alteraes
48
que podem ocorrer quando um mtodo transferido para outros laboratrios, analistas ou equipamentos (HEYDEN, 1994).
Para determinar a robustez de um mtodo, o INMETRO recomenda o teste de Youden (INMETRO, 2003). Trata-se de um teste que permite no s avaliar a robustez do mtodo, como tambm ordenar a influncia de cada uma das variaes nos resultados finais, indicando qual o tipo de influncia de cada uma dessas variaes. Por este teste so realizados oito ensaios com uma combinao fatorial dos efeitos e verifica-se qual o efeito ou combinao de efeitos que apresentam variaes.
A IUPAC (THOMPSON et al., 2002) utiliza o mesmo conceito de robustez para a palavra ruggedness. A USP tambm utiliza o termo ruggedness, mas com uma definio diferente, que lembra reprodutibilidade: A robustez de um mtodo analtico o nvel de reprodutibilidade dos resultados dos testes obtidos pelas anlises de algumas amostras sob uma variedade de condies normais de teste, tais como diferentes laboratrios, diferentes analistas, diferentes instrumentos, diferentes lotes de reagentes, diferentes dias, etc.(USP, 1999).
Em HPLC, a robustez pode ser avaliada, por exemplo, variando o contedo de metanol na fase mvel em 2%, o pH da fase mvel em 0,1 unidades de pH ou a temperatura da coluna em 5 C. Se estas mudanas estiverem dentro dos limites de exatido, e preciso e seletividade aceitveis, ento o mtodo possui robustez e tais variaes podem ser incorporadas ao procedimento. Em trabalhos nos quais h mudanas de fornecedores, marcas ou equipamentos ao longo do desenvolvimento e validao das metodologias, sem alterao significativa nos resultados, pode-se dizer que o mtodo possui uma robustez intrnseca, pois manteve sua resposta em meio a mudanas de ambiente de anlise.
49
6 ESTABILIDADE DOS PADRES E DAS AMOSTRAS
Para gerar resultados confiveis e reprodutveis, as amostras, os padres e reagentes usados devem ser estveis por um perodo razovel (por ex. um dia, uma semana, um ms, dependendo da necessidade) (SHABIR, 2003). Freqentemente, em equipamentos automatizados, as corridas cromatogrficas so realizadas durante a noite para melhor aproveitamento do funcionamento do laboratrio. Esta prtica requer maior estabilidade das solues. A estabilidade das amostras e padres importante em termos de temperatura e tempo. Se uma soluo no for estvel em temperaturas ambientes, a diminuio da temperatura pode aumentar a estabilidade das amostras e padres. Com relao ao tempo, estabilidade de dias ou meses mais desejvel, entretanto em alguns casos, as solues precisam ser preparadas cada vez que forem realizadas as anlises (SNYDER et al., 1997).
Em certos tipos de amostras, faz-se necessrio avaliar a estabilidade da substncia para determinar o tempo de estocagem das amostras (GREEN, 1996). Tempos longos de estocagem de amostras biolgicas, por exemplo, aumentam a probabilidade de degradao dos compostos de interesse, com subseqente formao de metablitos. Conhecendo a estabilidade, as anlises podem ser completadas antes de ocorrer a degradao.
7 CONFORMIDADE DO SISTEMA
Antes de realizar experimentos de validao ou mesmo anlises de amostras, deve-se avaliar se o sistema utilizado para a anlise capaz de fornecer dados de qualidade aceitvel. Esta avaliao alcanada com experimentos de conformidade do sistema (system suitability), que pode ser definida como um conjunto de testes para garantir que o equipamento utilizado est apto a gerar resultados de exatido e preciso aceitveis. A conformidade do sistema pode causar dvidas quanto ao seu alcance e, por isso, encontrada em duas abordagens.
50
A primeira considera que a resoluo e a repetitividade do sistema cromatogrfico sejam adequadas para a anlise a ser realizada. Assim, a conformidade do sistema verificada antes que o desenvolvimento do mtodo e a validao tenham sido completados. Os critrios selecionados so baseados no desempenho do mtodo determinado durante a validao. Por exemplo, se o tempo de reteno da amostra fizer parte do critrio de conformidade do sistema, a sua variao (estimativa do desvio padro) pode ser determinada durante a validao, que pode ter uma variao de 3%, por exemplo, (baseado nos resultados de validao) durante o uso rotineiro.
A segunda abordagem considera o sistema como um todo e inclui, alm do sistema cromatogrfico, a calibrao e manuteno dos equipamentos e instrumentos utilizados em todo o procedimento analtico, dentro das especificaes. Neste caso, a conformidade do sistema baseia-se no conceito de que o equipamento, componentes eletrnicos, operaes analticas e amostras constituem um sistema integral que pode ser avaliado como um todo. Pode-se dizer, ento, que a conformidade do sistema a verificao de um sistema para garantir a sua qualidade antes ou durante a anlise de amostras desconhecidas.
Alguns autores consideram que, se o sistema estiver qualificado, a validao do mtodo pode ser desenvolvida. Algumas vezes os critrios para avaliao da conformidade do sistema so definidos antes da validao e, quando realizados durante as anlises, estes testes garantem que o desempenho do sistema est apropriado para o uso.
Os parmetros a serem medidos e seus limites recomendados, de acordo com a US-FDA, esto no quadro 2 (FDA, 2000). Tipicamente, no mnimo dois destes critrios so requeridos para garantir a conformidade do sistema.
51
Quadro 2 Parmetros de conformidade do sistema e recomendaes. Parmetro Recomendao
O pico deve estar bem separado de outros picos e do pico correspondente ao tempo de reteno de um composto no retido (tM), k > 2 RSD < 1% para n > 5 Rs > 2 entre o pico de interesse e o interferente potencial mais prximo (impureza, produto de degradao) TF 2 Em geral deve ser > 2000 para HPLC
Fator de reteno (k)
Repetitividade (RSD) Resoluo (Rs) Fator de alargamento (TF) Nmero de pratos da coluna (N)
8 REVALIDAO Dentro de um laboratrio provvel que, aps um perodo de tempo, certos reagentes e equipamentos possam ter sofrido alteraes, seja por mudana de fornecedor, troca de componentes ou desgaste do equipamento provocado pelo uso constante. possvel que o desempenho do mtodo e, ento, a validade dos resultados gerados pelo mtodo sejam afetados por estas mudanas. A revalidao, que pode ser necessria em tal situao, a reavaliao de um mtodo analtico validado em resposta a uma mudana em algum aspecto do mtodo (JENKE, 1998; HILL; REYNOLDS, 1999). impraticvel e provavelmente desnecessrio revalidar um mtodo que tenha sofrido pequenas mudanas. Prope-se que estas pequenas variaes sejam avaliadas durante a validao, no parmetro de robustez, e que a revalidao de mtodo seja limitada s situaes como mudanas de maiores extenses. Para mtodos de separao, alteraes significativas poderiam ser devido a mudanas no produto para o qual o mtodo foi validado, no instrumento, no reagente (tipo ou fabricante) ou no procedimento.
A revalidao tambm deve ser considerada quando a proposta e/ou o nvel de qualidade desejado do mtodo so alterados, o procedimento modificado, ou mesmo quando um mtodo usado novamente aps certo perodo de tempo.
52
Com respeito as quais parmetros devem ser inclusos na revalidao, pode-se dizer que quanto maiores as alteraes no mtodo, maior deve ser a abrangncia da revalidao.
CAPTULO 2: Sistema de Deteco de Frmacos
1 CROMATOGRAFIA LQUIDA DE ALTA EFICINCIA (HPLC)
Entre os mtodos modernos de anlise, a cromatografia ocupa um lugar de destaque devido a sua facilidade em efetuar a separao, identificao e quantificao das espcies qumicas, por si mesma ou em conjunto com outras tcnicas instrumentais de anlise, como, por exemplo, a espectrofotometria ou a espectrometria de massa.
A cromatografia um mtodo fsico-qumico de separao dos componentes de uma mistura, realizada atravs da distribuio destes componentes entre duas fases, que esto em contato ntimo. Uma das fases permanece estacionria enquanto a outra se move atravs dela. Durante a passagem da fase mvel sobre a fase estacionria, os componentes da mistura so distribudos entre as duas fases, de tal forma que cada um dos componentes seletivamente retido pela fase estacionria, resultando em migraes diferenciais destes componentes (COLLINS et al., 1997).
Para o acompanhamento de frmacos in vivo so utilizadas metodologias analticas capazes de quantificar e identificar com preciso e exatido os mesmos em fludos biolgicos. Uma das tcnicas mais empregadas para este tipo de anlise a cromatografia lquida de alta eficincia (HPLC).
Na utilizao desta tcnica as amostras devem passar por um processo de purificao. Na rotina, as tcnicas mais utilizadas para extrao e/ou prconcentrao de compostos presentes no fludo biolgico so: extrao lquidolquido e extrao em fase slida (LOBO, 2001).
53
Para
uso
da
extrao
lquido-lquido
em
fluidos
biolgicos
primeiramente necessria a escolha adequada do solvente orgnico e ajuste do pH da amostra para que ocorra uma boa recuperao do analito. Vrios tipos de solventes orgnicos so utilizados para extrao de drogas cidas e bsicas presentes na amostra de fludos biolgicos. Quanto maior a afinidade do analito pelo solvente orgnico maior a recuperao. Substncias bsicas so normalmente extradas em pH maior que 7,0 e extrao de substncia cida em pH menor que 5,0. Este tipo de extrao muito utilizado para substncias presentes em fludos biolgicos (LOBO, 2001).
Esse tipo de extrao vantajoso por ser simples e pela variedade de solventes, puros e disponveis comercialmente, os quais fornecem uma ampla faixa de solubilidade e seletividade. Alm disso, ocorre a desnaturao das protenas presentes na amostra, eliminando, assim, a contaminao da coluna cromatogrfica (LOBO, 2001).
A extrao lquido-lquido possui algumas desvantagens: as amostras que possuem grande afinidade pela gua, que so parcialmente extradas pelo solvente orgnico, resultam em perda do analito. necessria a utilizao de solventes ultrapuros, uma vez que impurezas do solvente so concentradas junto com a amostra. O grande volume de solvente utilizado acaba gerando problemas de descartes, alm de ser um processo susceptvel a erros e de difcil automao. Apesar destas desvantagens, a extrao lquido-lquido considerada uma tcnica clssica de pr-tratamento de amostra.
As tcnicas de extrao e/ou pr-concentrao permitem que a anlise dos componentes de interesse se torne possvel. A meta final a obteno de uma frao da amostra original enriquecida com as substncias de interesse analtico, de forma que se obtenha uma separao cromatogrfica livre de interferentes, com deteco adequada e um tempo razovel de anlise (LOBO, 2001).
A cromatografia lquida de alta eficincia o mais importante membro de uma famlia inteira de tcnicas de separao. Utiliza instrumentos muito sofisticados
54
que podem ser totalmente automatizados. um tipo de cromatografia lquida que emprega pequenas colunas recheadas de materiais especialmente preparados e uma fase mvel que eluda sob altas presses. As colunas utilizadas so muito eficazes, porm oferecem grande resistncia vazo da fase mvel. Por esta razo, necessrio empregar sistemas de bomba de alta presso (at 400 bars) que fazem a fase mvel migrar a uma velocidade razovel atravs da coluna analtica (COLLINS et al., 1997).
A cromatografia lquida de alta eficincia (HPLC) tem a capacidade de realizar separaes e anlises quantitativas de uma grande quantidade de compostos presentes em vrios tipos de amostras, em escala de tempo de poucos minutos, com alta resoluo, eficincia e sensibilidade. A deteco contnua e com grande reprodutibilidade fornecida por esta tcnica eleva as anlises qualitativa e quantitativa a um alto nvel de exatido e preciso.
1.1 Detector por absorbncia no ultravioleta e no visvel
Nos detectores espectrofotomtricos o seu funcionamento baseia-se na absorbncia de luz por parte da amostra ao passar atravs dela qualquer radiao eletromagntica; normalmente isto ocorre no ultravioleta at o infravermelho, em um dado comprimento de onda. Existem dois tipos de detectores de luz ultravioleta: o de comprimento de onda varivel (espectrofotmetros), que no s de aplicao mais variada e sensvel, mas tambm mais caro, e o chamado fotomtrico que funciona com um ou dois comprimentos de onda fixos. Este ltimo sensvel, econmico e mais que suficiente para se conseguir bons resultados com todos os compostos que absorvem luz no comprimento de onda em que ele funciona. Este tipo de detector normalmente insensvel a variaes de vazo e temperatura. A maioria dos detectores de comprimento de onda fixo, oferecidos no mercado, operam em um comprimento de onda de 254 nm e um de 280 nm, resultado da absorbncia de luz em 254 nm e da emisso de luz em 280 nm por uma substncia fosforescente. Em timas condies, pode-se atingir sensibilidades de at 0,001 unidades de absorbncia e, se o composto absorve intensamente na faixa de UV, possvel
55
detectar quantidades de amostras da ordem de nanogramas (10-9 g) (SKOOG; LEARY, 1992; SNYDER et al., 1997).
Espectrofotmetros de comprimentos de onda varivel UV-VIS, cobrindo a faixa de 190-800 nm, atravs de monocromador que seleciona o comprimento de onda desejado do feixe de luz emitido pelas lmpadas de deutrio (UV) ou tungstnio (VIS), oferecem vrias vantagens sobre os instrumentos de comprimento de onda fixo: a) Apresenta alta absorbncia para vrios componentes devido escolha de comprimento de onda e, conseqentemente, tem maior sensibilidade. b) Permite maior seletividade, desde que um determinado comprimento de onda pode ser escolhido, onde o soluto de interesse absorve bastante e outros no. c) Promove eficincia em eluio por gradiente atravs da habilidade de selecionar um comprimento de onda onde os componentes da fase mvel no apresentam uma variao de absorbncia para diferentes concentraes. d) Permitir obter o espectro de absorbncia de cada componente em separado aps parar a vazo da fase mvel. Anlises em diferentes comprimentos de onda tambm so possveis com os detectores espectrofotomtricos atravs de um conjunto de fotodiodos. Esses fotodiodos, seletivos a determinados comprimentos de onda, em grande nmero, cerca de 250, permitem levantar o espectro da substncia durante a eluio. Este detector denominado detector de rede de diodos (diode array detector). Os detectores de rede de diodos eram, at a alguns anos, pouco sensveis, mas na atualidade, com a melhora dos computadores com programas dedicados para este fim, com o aumento da sensibilidade dos diodo se com correes da aberrao da rede de difrao, eles aumentaram sua sensibilidade e aplicabilidade (SKOOG; LEARY, 1992; SNYDER et al., 1997).
56
Figura 3. Aparelho de cromatografia lquida de alta eficincia com deteco de ultravioleta utilizado na quantificao das amostras contendo amoxicilina.
1.2 Detector por Fluorescncia
A espectroscopia de fluorescncia um mtodo de deteco dos mais sensveis da atualidade, especfico para compostos que fluorescem. Em boas condies possvel detectar quantidades da ordem de picogramas (10-12 g), o que comparvel aos detectores por captura de eltrons. Uma alta intensidade de fluorescncia esperada de compostos que sejam conjugados simetricamente ou que no podem produzir estruturas fortemente inicas.
A fase mvel empregada nos detectores de fluorescncia deve ser cuidadosamente selecionada, pois a intensidade de emisso depende do meio em que se encontra a amostra. Isto dificulta algumas de suas aplicaes, tais como anlise quantitativa e eluio por gradiente, nas quais se devem selecionar os componentes da fase mvel para no atrapalhar a fluorescncia. Os detectores para fluorescncia tambm podem proporcionar espectros de emisso das amostras retidas momentaneamente na sua cela. Desde que uma fonte de UV necessria em ambos os detectores, por absorbncia no UV e por fluorescncia, eles podem
57
ser combinados em um s detector (SKOOG; LEARY, 1992; SNYDER et al., 1997; LEENHEER et al., 1988).
Figura 4. Aparelho de cromatografia lquida de alta eficincia com deteco de fluorescncia utilizado na quantificao das amostras contendo norfloxacino.
1.3 Detector por Espectrometria de Massa
A espectrometria de massa uma tcnica analtica poderosa usada para identificar compostos desconhecidos, quantificar materiais conhecidos e elucidar as propriedades qumicas e estruturais das molculas. A deteco de compostos pode ser conseguida para quantidades to pequenas como 10-15 g para um composto de massa de 1000 Dalton. Isto significa que os compostos podem ser identificados em concentraes muito baixas (da ordem de 10-12) em misturas quimicamente complexas. A espectrometria de massa fornece informao tanto para qumicos, quanto para fsicos, engenheiros, bioqumicos e bilogos, entre outros profissionais. Os princpios cientficos em que a tcnica se baseia so simples. A essncia da tcnica envolve a gerao de ons que so depois detectados. A sofisticao surge nos mtodos que so usados para a gerao desses mesmos ons e no modo de analis-los (figura 5).
58
Figura 5. Esquema de funcionamento do sistema de espectrometria de massa. O desenvolvimento da espectrometria de massa de ionizao por eletrospray (IES) permitiu novas possibilidades para anlise de compostos de elevada massa molecular de todos os tipos, incluindo protenas, nucleotdeos e polmeros sintticos, sendo por isso uma tcnica muito usada em diversas reas de pesquisa.
Comparvel ao excitante desenvolvimento da ressonncia magntica nuclear durante as ltimas trs dcadas, a espectrometria de massa entrou em uma fase de crescimento acelerado nos anos oitenta, comeando com a introduo de mtodos de ionizao. incrvel a velocidade com a qual a espectrometria de massa tem se expandido dentro de reas de pesquisa, nas quais, tradicionalmente, era limitado o seu uso (SIUZDAK, 2004).
A cromatografia gasosa associada espectrometria de massa (GC/MS) tem sido considerada uma tcnica analtica adequada para a anlise de misturas complexas. Tem, no entanto, a grande limitao de ser aplicvel apenas a molculas relativamente volteis e termicamente estveis. Um acoplamento semelhante entre a cromatografia lquida e a espectrometria de massa (LC/MS) era, por isso, de todo o interesse, para a anlise de compostos sem essas caractersticas, para os quais a anlise por GC/MS s podia ser utilizada recorrendo a derivaes que tornam o processo analtico muito demorado.
59
A IES atravs da espectrometria de massa sofreu um rpido crescimento, tornando-se, assim, uma tcnica analtica fundamental para anlise de uma vasta gama de compostos polares, no volteis e termicamente instveis, indo de compostos de baixa massa molecular at biopolmeros de elevada massa molecular.
As principais caractersticas dos espectros de massa de biomolculas so as predominncias de ons moleculares multiplamente carregados e as ausncias de fragmentao que permitem a determinao rigorosa de massas moleculares. No entanto, no que diz respeito estrutura molecular, pouca informao, em geral, obtida. A dissociao pode ainda ser provocada por ativao colisional na regio de decomposio de um espectrmetro acoplado a outro (MS/MS), sendo esta tcnica relevante na obteno de informao estrutural de molculas. Uma mistura de compostos introduzida e, com o primeiro espectrmetro de massa, seleciona-se um on de interesse. Este depois dissociado entre os dois espectrmetros, geralmente por coliso com gs argnio a certa presso, transformando a energia cintica em energia vibracional. O segundo espectrmetro separa e determina os valores de m/z dos ons fragmentados para posterior deteco (ANTIGNAC et al., 2000). A figura 6 e 7 mostra o aparelho e o esquema da fragmentao utilizando espectrmetro de massa no modo de monitoramento de reao mltipla (MRM).
Figura 6. Aparelho de cromatografia lquida de alta eficincia acoplado ao espectrmetro de massa utilizado na quantificao das amostras contendo oxcarbazepina.
60
Mistura de compostos
Seleo do on de interesse
Dissociao
Seleo do on fragmento de interesse
FONTE
Q1
Q2
Q3
DETECTOR
Figura 7. Esquema demonstrativo da fragmentao de compostos utilizando espectrmetro de massa triplo-quadrupolo no modo de monitoramento de reao mltipla (MRM).
CAPTULO 3: Estudos Farmacocinticos e Bioequivalncia
1 ASPECTOS QUALITATIVOS
Quando um frmaco introduzido num organismo vivo, este apresenta propriedades farmacodinmicas e farmacocinticas. A farmacodinmica estuda o efeito desses compostos no organismo bem como os mecanismos atravs dos quais elas atuam. A farmacocintica a parte da farmacologia encarregada de estudar os fenmenos envolvidos nos processos de absoro, distribuio, metabolismo e excreo de um medicamento (PAGE et al., 2002).
A absoro, a distribuio, a biotransformao e a eliminao de uma substncia envolvem a sua passagem atravs das membranas celulares. As caractersticas mais importantes de um frmaco so sua estrutura e massa molecular, sua solubilidade no local de absoro, o grau de ionizao e a lipossolubilidade relativa de suas formas ionizadas e no-ionizadas (GOODMAN; GILMAN, 2007).
Quando uma substncia penetra numa clula, precisa atravessar a membrana plasmtica celular, na qual as molculas proticas globulares tambm
61
penetram ou a atravessam por completo uma dupla camada fluida de fosfolipdio (SINGER; NICHOLSON, 1972).
Cada molcula de lipdio, na camada dupla, consegue mover-se lateralmente conferindo membrana fluidez, flexibilidade, elevada resistncia eltrica e impermeabilidade relativa s molculas extremamente polares. Complexos de protenas intrnsecas da membrana e lipdios conseguem formar canais hidrofbicos e hidroflicos que permitem o transporte de molculas com caractersticas diferentes (RIDOUT et al., 1988).
Os frmacos atravessam as membranas tanto atravs de processos passivos como por mecanismos que envolvem a participao ativa dos componentes da membrana. No primeiro caso, a molcula do frmaco costuma atravessar por difuso passiva atravs de um gradiente de concentrao graas sua solubilidade na camada lipdica. Esta transferncia diretamente proporcional magnitude dos gradientes de concentrao atravs da membrana e a lipossolubilidade do frmaco. Quanto mais lipossolvel, maior a concentrao do frmaco na membrana e mais rpida a sua difuso. Aps ter atingido um estado de equilbrio dinmico, a concentrao de frmaco livre igual de ambos os lados da membrana. No caso de compostos inicos, as concentraes no estado de equilbrio dinmico dependero de diferenas de pH atravs da membrana, influenciam a ionizao da molcula de cada lado da membrana e o gradiente eletroqumico do on. A maioria das membranas biolgicas relativamente permevel gua, seja por difuso ou pelo fluxo que resulta de diferenas hidrostticas ou osmticas atravs da membrana. Este fluxo de gua pode carrear consigo pequenas substncias hidrossolveis. De modo geral, essas substncias no atravessam as membranas celulares quando suas massas moleculares so superiores a 100 m (BRODIE, 1964; PRESCOTT, 1981).
1.1 Absoro de Frmacos
A absoro descreve a velocidade com a qual o frmaco deixa seu local de administrao e a magnitude com que isso ocorre. Muitas variveis, alm dos
62
fatores fsico-qumicos que afetam o transporte atravs das membranas, influenciam a absoro dos frmacos (GOODMAN; GILMAN, 2007).
As substncias administradas em soluo aquosa so absorvidas mais rapidamente que aquelas administradas em solues oleosas, suspenses ou forma slida; porque se misturam mais facilmente com a fase aquosa no local de absoro. No caso de substncias administradas na forma slida, a velocidade de dissoluo ser o fator limitante da sua absoro, como representado na figura 8 (PRESCOTT, 1981).
Figura 8. Processo de absoro de formas farmacuticas slidas.
A concentrao de um frmaco influncia a velocidade de sua absoro. Os agentes ingeridos ou injetados em solues de concentrao elevada so absorvidos mais rapidamente do que aqueles em soluo de baixa concentrao. A circulao para o local da absoro tambm afeta a absoro do frmaco. O maior fluxo sanguneo aumenta a velocidade de absoro do frmaco, enquanto a reduo do fluxo sanguneo diminui a absoro (PRESCOTT, 1981).
A rea de superfcie absorvente qual um frmaco exposto um dos determinantes mais importantes da velocidade de absoro. As substncias so absorvidas muito rapidamente a partir de grandes superfcies, como a mucosa intestinal. Como a maior parte da absoro do trato gastrointestinal ocorre atravs de processos passivos, a absoro favorecida quando o frmaco se encontra na forma no-ionizada, que mais lipoflica (BRODIE, 1964).
63
1.2 Distribuio de Frmacos
Aps ser absorvido ou injetado na corrente sangunea, o frmaco distribui-se para os lquidos intersticiais e celulares. Pode ser diferenciada uma fase inicial de distribuio que reflete o dbito cardaco e o fluxo sanguneo regional. O corao, o fgado, os rins, o crebro e outros rgos bem perfundidos recebem maior parte dos frmacos durante os primeiro minutos aps sua absoro. O aporte do frmaco musculatura, maioria das vceras, pele e ao tecido adiposo mais lento, e estes tecidos exigem de minutos a algumas horas para atingir o estado de equilbrio, como representado na figura 9 (BENET, 1978).
Figura 9. Distribuio de frmacos no organismo.
Uma segunda fase de distribuio do frmaco pode ento ser percebida e est tambm limitada pelo fluxo sanguneo; envolvendo uma frao muito maior da massa corporal do que a primeira. Os frmacos, que no so lipossolveis e penetram mal nas membranas tm sua distribuio limitada e, portanto, seus locais de ao potenciais tambm limitados. Alm disso, a distribuio pode ser limitada por uma ligao dos frmacos s protenas plasmticas, sobretudo albumina (no caso dos frmacos cidos) e glicoprotena cida (no caso de substncias bsicas). Um agente que apresenta ligao extensa e forte com as protenas plasmticas tem acesso limitado aos locais de ao celulares e metabolizado e eliminado lentamente. Os frmacos podem acumular-se nos tecidos em concentraes
64
maiores do que o esperado nos equilbrios de difuso, como resultado do gradiente de pH, ligao com componentes intracelulares ou distribuio em lipdios (ROBERTS et al., 1988). Os frmacos que se acumulam em determinado tecido podem servir como reservatrio que prolonga suas aes no mesmo tecido ou num local distante atingido atravs da circulao (NEBERT; GOZALEZ, 1987).
Uma terceira fase de distribuio destes frmacos origina-se da lenta captao do frmaco pelo tecido adiposo (limitada pelo fluxo sanguneo). Esses pontos podem tornar-se reservatrios para a manuteno da concentrao plasmtica (BENET, 1978).
1.3 Biotransformao de Frmacos
A biotransformao enzimtica de frmacos em metablitos mais polares e menos lipossolveis aumenta sua excreo e reduz seu volume de distribuio. Tal biotransformao alivia a carga de substncias qumicas estranhas e essencial sobrevida do organismo (GOLDSTEIN et al., 1974).
Os sistemas enzimticos responsveis pela biotransformao de muitos frmacos esto localizados no retculo endoplasmtico liso dos hepatcitos no fgado (frao microssmica). Tais enzimas tambm so encontradas em outros rgos como rins, pulmes e epitlio gastrointestinal. Os frmacos absorvidos pelo intestino esto, portanto, sujeitos ao efeito de primeira passagem. Isto representa a ao combinada de enzimas epiteliais gastrointestinais e hepticas, que algumas vezes podem impedir que concentraes efetivas do frmaco ativo atinjam a circulao sistmica aps a administrao oral, como j foi comentado antes (NEBERT; GOZALEZ, 1987).
As reaes qumicas da biotransformao enzimtica so classificadas como reaes em fase I ou II (figura 10). As reaes de fase I convertem o frmaco original em um metablito mais polar atravs de oxidao, reduo ou hidrlise. O metablito resultante pode ser farmacologicamente inativo, menos ativo ou mais ativo que a molcula original. Quando o prprio metablito a forma ativa, o
65
composto original denominado de pr-frmaco. As reaes de fase II, que tambm so denominadas reaes de conjugao ou de sntese, envolvem a ligao do frmaco ou de seu metablito polar a um substrato endgeno como glicuronato, sulfato, acetato ou um aminocido (PAGE et al., 2002).
Reaes Fase I Reaes Fase II
- Oxidao - Reduo - Hidrlise
- Conjugao com cido glucurnico - Conjugao com peptdeos - Conjugao com sulfatos - Metilao - Acetilao - Sntese de cido mercaptrico
Figura 10. Biotransformao de frmacos.
Os estudos de biotransformao em animais de laboratrio mostram que um grande nmero de fatores genticos, ambientais e fisiolgicos afeta a metabolizao de um frmaco. No homem, os fatores mais importantes so polimorfismos geneticamente determinados nas oxidaes e conjugaes dos frmacos, influncias ambientais, incluindo uso concomitante de outras substncias que induzam ou inibam enzimas metabolizadoras de frmacos, e a existncia de doenas hepticas, quase sempre associadas grave desnutrio (BRIDGES, 1987).
1.4 Eliminao de frmacos
Os frmacos so eliminados do organismo tanto na forma inalterada como na forma de metablitos. Os rgos excretores, com a excluso dos pulmes, eliminam os compostos polares de forma mais eficientes do que as substncias com lipossolubilidade elevada. Os frmacos lipossolveis, portanto, no so eliminados at serem metabolizados em compostos mais polares. O rim o rgo mais importante de eliminao de frmacos e metablitos (GUYTON, 1973).
66
As
substncias
eliminadas
nas
fezes
so,
sobretudo,
aquelas
administradas por via oral e que no so absorvidas ou metablitos excretados na bile que no so reabsorvidos pelo trato gastrointestinal. A excreo pulmonar importante, sobretudo, para a eliminao de vapores e gases anestsicos e, s vezes, pequenas concentraes de outros frmacos ou metablitos so eliminados por esta via (GUYTON, 1973). A eliminao de frmacos e metablitos na urina envolve trs processos: filtrao glomerular, secreo ativa e reabsoro tubular passiva (ATKINSON, 1988).
A concentrao do frmaco que penetra na luz tubular por filtrao depende da sua ligao protica plasmtica fracional e da velocidade de filtrao glomerular. No tbulo proximal, determinados nions e ctions orgnicos so adicionados ao filtrado glomerular atravs de secreo tubular ativa mediada por carreadores (ATKINSON et al., 2001; GUYTON, 1973).
Os sistemas carreadores so relativamente no seletivos e ons orgnicos de carga eltrica semelhante competem pelo transporte. Os sistemas de transporte tambm podem ser bidirecionais e pelo menos alguns frmacos so secretados e ativamente reabsorvidos. Todavia, o transporte da maioria dos ons exgenos predominante secretrio. Nos tbulos proximais e distais, as formas no ionizadas de cidos e bases fracos sofrem reabsoro passiva final. O gradiente de concentrao da retrodifuso criado pela reabsoro da gua com Na+ e outros ons inorgnicos. Como as clulas tubulares so menos permeveis s formas ionizadas que ao eletrlito fraco, a reabsoro passiva dessas substncias pH dependente. Quando a urina tubular se torna mais alcalina, os cidos so eliminados mais rapidamente, basicamente porque esto mais ionizados e diminuem a reabsoro passiva. Quando a urina tubular acidificada, diminui a excreo de cidos fracos. A alcalinizao e a acidificao da urina tm efeitos opostos sobre a excreo das bases fracas (GOODMAN; GILMAN, 2007).
Muitos metablitos formados no fgado so eliminados na bile para o trato intestinal. Esses metablitos podem ser excretados nas fezes, reabsorvidos para o sangue e por fim eliminados na urina. A excreo de frmacos pelo suor, saliva,
67
pelas lgrimas e leite materno no importante do ponto de vista quantitativo (ATKINSON et al., 1988).
2 ASPECTOS QUANTITATIVOS
Uma hiptese fundamental da farmacocintica clnica que existe uma correlao entre a resposta farmacolgica ou txica a um frmaco e a sua concentrao na circulao sistmica e que est relacionada sua concentrao nos seus locais de ao (PAGE et al., 2002; ROWLAND; TOZER, 1995).
As inmeras variveis fisiolgicas e fisiopatolgicas que ditam os ajustes posolgicos em pacientes, freqentemente o fazem como resultado de uma modificao dos parmetros farmacocinticos. Os trs parmetros mais importantes so: depurao (uma medida da capacidade do organismo de eliminar o frmaco), volume de distribuio (uma medida no espao aparente do corpo disponvel para conter o frmaco) e biodisponibilidade (a frao do frmaco absorvida como tal na circulao sistmica) (ATKINSON et al., 1988; SHORGET; ANDREW, 1941).
O conceito de depurao (Clearence CL) extremamente valioso na famacocintica clnica, porque a eliminao de determinado frmaco costuma ser constante na amplitude de concentraes encontradas clinicamente. Em nveis simples, a depurao de um frmaco a taxa de eliminao por todas as vias normalizadas de acordo com a concentrao do frmaco (C) em algum lquido biolgico (KLOTZ et al., 1975; ROWLAND; TOZER, 1995):
CL = Taxa de eliminao / C
importante observar que a depurao no indica quanto do frmaco est sendo eliminado, mas sim o volume de lquido biolgico, como o sangue e o plasma, que precisa estar completamente livre do agente para contribuir para sua eliminao. A depurao expressa como volume por unidade de tempo. Em geral, a depurao costuma ser definida como depurao sangunea (CLb), depurao plasmtica (CLp) ou depurao baseada na concentrao de frmaco livre (CLu),
68
dependendo da concentrao medida (Cb, Cp ou Cu) (BENET, 1984; ATKINSON et al., 2001).
A depurao atravs de vrios rgos de eliminao aditiva. A eliminao do frmaco pode ser decorrente dos processos que ocorrem nos rins, no fgado e em outros rgos. A diviso da taxa de eliminao por cada rgo por uma concentrao do frmaco (concentrao plasmtica) fornecer a depurao respectiva deste rgo. Em conjunto, tais depuraes separadas sero iguais depurao sistmica total (GOODMAN; GILMAN, 2007; SHORGET; ANDREW, 1941; ROWLAND; TOZER, 1995):
CLrenal + CLheptica + CLoutros = CLsistmica
Para uma nica dose de um frmaco com biodisponibilidade completa e cintica de eliminao de primeira ordem, a depurao sistmica total poderia ser (KLOTZ et al., 1975):
CL = Dose/AUC
Na qual AUC a rea total sob a curva que descreve a concentrao do frmaco na circulao sistmica em funo do tempo (de zero at o infinito) (KLOTZ et al., 1975).
O volume um segundo parmetro fundamental na discusso dos processos de distribuio farmacolgica. O volume de distribuio (V) relaciona a concentrao de frmaco no organismo com a concentrao de frmaco (C) no sangue e no plasma, dependendo do lquido medido. Este volume no se refere necessariamente a um volume fisiolgico identificvel, mas apenas ao volume de lquido que seria necessrio para conter todo o frmaco no organismo, na mesma concentrao em que se encontra no sangue ou no plasma (BENET; GALEAZZI, 1979; ROWLAND; TOZER, 1995):
69
O volume de plasma de um homem normal de 70 kg de 3 litros, o volume sanguneo de aproximadamente 5,5 litros, o volume de lquido extracelular fora do plasma de 12 litros e o volume de gua corporal total de aproximadamente 42 litros. Entretanto, muitos frmacos exibem volumes de distribuio muito superiores a estes valores. No caso de substncias que apresentam ligao substancial com as protenas plasmticas, mas que no se ligam aos componentes teciduais, o volume de distribuio aproximar-se- do volume plasmtico. Em contrapartida, determinados frmacos apresentam volume de distribuio elevado, embora a maior parte do frmaco na circulao esteja ligada albumina, porque tais frmacos tambm so seqestrados em outros pontos do organismo (BENET et al., 1984; SHORGET; ANDREW, 1941).
A meia-vida de um frmaco o tempo que leva para a concentrao plasmtica ou a concentrao da substncia no corpo reduzir em 50%. No caso mais simples, o modelo monocompartimental, a meia-vida pode ser determinada rapidamente e usada para tomar decises sobre posologia do frmaco (BENET, 1984).
A meia-vida um parmetro que varia de acordo com a depurao e o volume de distribuio. Uma correlao aproximada entre a meia-vida clinicamente importante, a depurao e o volume de distribuio dada por (GOODMAN; GILMAN, 2007; ROWLAND; TOZER, 1995):
t1/2= 0,693 x V/CL
A depurao a capacidade do organismo em eliminar uma substncia. Entretanto, os rgos de eliminao s podem retirar o frmaco do sangue ou do plasma com o qual esto em contato direto (ATKINSON et al., 2001; SHORGET; ANDREW, 1941). Embora possa ser um indicador satisfatrio de eliminao do frmaco, a meia-vida um bom indicador do tempo necessrio para atingir um estado de equilbrio dinmico (KLOTZ et al., 1975).
70
2.1 Modelos Farmacocinticos
Uma hiptese ou modelo concebida usando termos matemticos, os quais so um meio conciso de expressar ralaes quantitativas. Vrios modelos matemticos podem ser usados para simular a velocidade ou taxa dos processos de absoro, distribuio e eliminao, sendo denominados, modelos farmacocinticos. Estes modelos possibilitam o desenvolvimento de equaes para descrever concentraes do frmaco no organismo em funo do tempo, as quais permitem caracterizar com reprodutibilidade o ambiente e o destino de um frmaco no sistema biolgico, aps sua administrao por uma determinada via de administrao e forma farmacutica (ROWLAND; TOZER, 1995).
Os parmetros farmacocinticos como Vd, t1/2 e clearance, so determinados experimentalmente a partir de curvas de concentrao (varivel dependente) em funo do tempo (varivel independente). Porm, para interpretao destas curvas e obteno dos parmetros, um modelo farmacocintico estimado e testado quanto a validade a partir destas; e uma vez validado os parmetros farmacocinticos so obtidos. Programas computacionais podem ser usados para estimar parmetros a partir de modelos farmacocinticos complexos. Devemos sempre lembrar que na verdade, como acabamos de ver, os parmetros farmacocinticos so adaptados de modelos e, por isto, so sujeitos a erros de estimativa que podem variar com as concentraes obtidas, tcnicas analticas de dosagens e metodologias utilizadas na interpretao dos dados (ROWLAND; TOZER, 1995).
2.1.1 Modelos compartimentais
O corpo pode ser representado como uma srie, ou sistemas, de compartimentos que comunicam-se reversivelmente entre si. Um compartimento no uma regio anatmica ou fisiolgica real, mas considerada como um tecido ou grupo de tecidos que devem possuir fluxo sangneo e afinidade pelo frmaco similar. Dentro de cada compartimento considera-se que o frmaco distribua-se
71
uniformemente; a mistura do frmaco dentro do compartimento rpida e homognea, tanto que sua concentrao representada como uma concentrao mdia e cada molcula do frmaco possui igual probabilidade de sair do compartimento. Modelos compartimentais so baseados em hipteses lineares usando equaes diferenciais e, embora os compartimentos farmacocinticos no corresponda a nenhuma das entidades anatmicas atuais, o compartimento, todavia, apresenta dimenses numricas de volume (ml, litro) como se fossem um volume real (ROWLAND; TOZER, 1995).
O emprego de modelos compartimentais em farmacocintica leva, geralmente, implcita a suposio de que os processos que se estudam, ou o fluxo dos frmacos at o compartimento, se desenvolvem segundo cintica de primeira ordem. Por exemplo, o resumo dos processos que retiram medicamentos do organismo irreversivelmente, pode caracterizar-se por uma constante de velocidade de eliminao de primeira ordem cintica, que compreenderia a excreo urinria, a biotransformao e outros processos que contribuem na retirada do frmaco do organismo (ROWLAND; TOZER, 1995).
A cintica de primeira ordem implica que a velocidade na qual se produz um processo proporcional quantidade ou concentrao do frmaco existente no compartimento no qual se desenvolve. Assim, se grande a quantidade de medicamento no organismo tambm , ou ser, alta a velocidade de eliminao. Por outro lado, a eliminao diminuir proporcionalmente com a reduo da quantidade ou concentrao (ROWLAND; TOZER, 1995).
Tambm a transferncia do medicamento de um compartimento a outro pode obedecer cintica de primeira ordem e o mesmo ocorre com a maioria dos processos que os frmacos experimentam no organismo. Em geral, alguns deles no so estritamente de primeira ordem, como por exemplo a biotransformao a secreo tubular ou a transferncia atravs de uma membrana quando processos ativos esto envolvidos. Estes podem obedecer uma cintica mais complexa, por exemplo a processos enzimticos regidos pela equao de Michaelis-Menten. No entanto, as concentraes de frmaco com as quais normalmente se trabalha em
72
farmacocintica (teraputicas), aparecem na maioria das vezes como de primeira ordem (ROWLAND; TOZER, 1995).
Assim, podemos dizer que os processos farmacocinticos correspondem a uma cintica linear. Uma conseqncia desta linearidade o fato de que a rea sob a curva (ASC) de concentrao plasmtica vs tempo, aps injeo por via intravenosa, uma funo linear da dose administrada (ROWLAND; TOZER, 1995).
Os modelos compartimentais consistem de um ou mais compartimentos perifricos conectados um compartimento central. O compartimento central representado pelo plasma e tecidos altamente perfundidos. Assim, quando uma dose intravenosa do frmaco administrada, ela entra diretamente no compartimento central e tambm deste compartimento que ocorre sua eliminao, uma vez que no compartimento central que se encontra os rgos envolvidos na eliminao, primariamente, rim, fgado e tecidos altamente perfundidos (ROWLAND; TOZER, 1995).
2.1.2 Modelo aberto de um compartimento
o modelo compartimental mais simples e pode representar frmacos que aps administrao, se distribuem atravs da via circulatria para todos os tecidos e se equilibra rapidamente em todo o organismo. Esta administrao pode ser na forma de injeo intravenosa rpida (IV bolus), atravs da qual toda a dose do frmaco entra imediatamente no organismo e, portanto, a velocidade de absoro negligenciada nos clculos; ou ainda por via extravenosa (VEV), onde a etapa de absoro deve ser considerada (ROWLAND; TOZER, 1995).
modelo
de
monocompartimental
descreve,
muitas
vezes
adequadamente, as alteraes sofridas ao longo do tempo, na concentrao plasmtica ou na excreo urinria de frmacos que aps a administrao, se distribuem rapidamente entre o plasma e os tecidos. Admitir a existncia de tal modelo no implica, necessariamente presumir que as concentraes plasmtica e tissular do frmaco sejam as mesmas, porm essencial que as alteraes que
73
ocorrem no plasma reflitam aquelas nos nveis tissulares do frmaco, ou seja que exista uma relao constante entre estas duas variveis (ROWLAND; TOZER, 1995).
2.1.3 Modelos multicompartimentais
Estes modelos so necessrios para explicar a observao de que aps uma rpida administrao IV a curva de nvel plasmtico vs tempo no declina linearmente como uma nica velocidade de primeira ordem. Em um modelo multicompartimental o frmaco se distribui a vrias velocidades dentro de diferentes grupos de tecidos. Aqueles que apresentam elevado fluxo sangneo podem equilibrar-se com o compartimento plasmtico, assim somado ao sangue compem o compartimento central. Enquanto esta distribuio inicial do frmaco efetuada, o frmaco liberado para um ou mais compartimentos perifricos compostos de grupos de tecidos com menor fluxo sangneo e afinidade pelo frmaco. Esta diferena que leva aparncia no linear da curva de concentrao sangnea do frmaco em escala logartmica vs tempo. Aps equilbrio do frmaco nestes tecidos perifricos a curva reflete, ento, eliminao de primeira ordem do frmaco para fora do organismo (ROWLAND; TOZER, 1995).
Com
objetivo
de
aplicar
anlise
cintica
em
modelos
multicompartimentais, devemos assumir que a velocidade geral do processo de passagem do frmaco entre os compartimentos de primeira ordem. Com base nesta suposio a curva de nvel plasmtico por tempo para um frmaco que segue um modelo multicompartimental melhor descrito pelo somatrio de vrios processos com velocidade de primeira ordem (ROWLAND; TOZER, 1995).
2.1.4 Modelo de dois compartimentos
Apesar extremamente til para muitos objetivos, o modelo de um compartimento muitas vezes no se aplica ao perfil do movimento de frmacos pelo organismo. A maioria destes casos pode ser resolvida aplicando-se um modelo um
74
pouco mais complexo, porm mais realstico, o de dois compartimentos (ROWLAND; TOZER, 1995).
Neste quando o frmaco introduzido diretamente no compartimento central (VIV) o nvel sangneo cai de maneira bifsica. A rpida queda inicial representa a distribuio do frmaco do compartimento central para o perifrico, embora sua eliminao comece a ocorrer desde que introduzido no organismo. Num certo momento, atingido um "pseudo-equilbrio" de distribuio entre o central e compartimento perifrico; isto ocorre quando a razo do frmaco entre os compartimentos se aproxima de um valor constante, mantido durante a segunda fase, mais lenta do declnio da concentrao sangnea do frmaco, a qual reflete principalmente sua eliminao. Durante essa segunda fase, a perda do frmaco pelo organismo descrita por um processo monoexponencial indicativo da homogeneidade cintica entre os nveis do frmaco em todos os lquidos e tecidos do organismo (ROWLAND; TOZER, 1995).
2.1.5 Modelos no compartimentais
Como sabemos os modelos compartimentais so uma simplificao do organismo e por isto deve ser aplicado com cautela. Alm disso esses modelos so altamente dependentes da espcie e, embora tenham muitos usos clnicos, a quantidade de informaes bsicas fornecidas, limitada; isto especialmente verdadeiro para a previso de nveis tissulares. Os modelos compartimentais no levam em conta a ligao frmaco-protena, embora possa ser afetado por ela (ROWLAND; TOZER, 1995).
Devido todas as limitaes referidas dos modelos compartimentais e visando a obteno de dados cada vez mais fidedignos modelos no compartimentais tem sido desenvolvidos e avaliados.
Estes
modelos
so
anatmica
fisiologicamente
realistas
desenvolvidos com base nos fluxos sangneos e volumes reais dos rgos, levando-se em conta tanto o frmaco ligado quanto o livre, no sangue ou tecidos.
75
Assim, descrevem mais realisticamente a disposio do frmaco em cada tecido ou rgo, no entanto, estes modelos so demasiadamente complexos nvel matemtico, perdendo universalidade (ROWLAND; TOZER, 1995).
A principal vantagem destes modelos a possibilidade de prever o comportamento farmacocintico de um determinado frmaco no homem, a partir de dados obtidos em animais e adaptados matematicamente para tal aplicao.
3 APLICAES DOS ESTUDOS FARMACOCINTICOS
O comportamento farmacocintico de um novo frmaco , normalmente, determinado em animais antes de ser utilizados em humanos. Os testes em animais de laboratrio seguem at o ponto em que se faz necessrio obter resultados em experimentos clnicos envolvendo seres humanos, pois existem variaes nos parmetros farmacocinticos quando comparamos resultados obtidos em animais e em humanos (ROBERTS et al., 1988).
Um nmero cada vez maior de novos frmacos com potencial teraputico, bem como a comercializao de produtos similares por diferentes empresas, tm estimulado o desenvolvimento de novos mtodos para o controle de qualidade. Os ensaios clnicos tornaram-se importantes ferramentas para avaliar isolada ou de forma comparativa as propriedades farmacocinticas e farmacodinmicas (ATKINSON et al., 2001).
O desenvolvimento e a regulamentao destes ensaios clnicos tornaramse reconhecidamente mais importante a partir da dcada de 50, quando passaram a ser conduzidos de acordo com os interesses cientficos e ticos (ATKINSON et al., 2001). Entretanto, foi em 1753 que Lind planejou e desenvolveu um dos primeiros ensaios clnicos de que se tem notcia. Em seu trabalho, utilizou 12 pacientes portadores de escorbuto, dividindo-os em grupos e tratamentos diferentes. Louis tambm executou importante estudo, demonstrando que a terapia de sangria, quando efetuada para tratamento de pneumonia (78 casos), erisipela (33 casos) e inflamao de garganta (23 casos), no causa qualquer benefcio quando
76
comparado com o grupo controle. Este resultado foi fundamental para o fim da tal prtica (LOUIS, 1834). Sutton realizou o primeiro trabalho descrito, com o uso de placebo como controle e demonstrou a tendncia natural de cura em alguns casos de febre reumtica (SUTTON, 1865). Holmes indicou a necessidade de progressos nas pesquisas clnicas dos Estados Unidos, para diminuir a incidncia de automedicao, num pblico que insiste em ser envenenado (HOLMES, 1891). Em 1915, Greenwood e Yule demonstraram a importncia da distribuio dos pacientes em grupo para a avaliao de resultados (GREENWOOD; YULE, 1915). Fergusson et al., em 1927, foram provavelmente os primeiros a introduzir o conceito de cegos aos estudos clnicos, fazendo testes em vacinas.
Na dcada de 30, as principais reas geradoras de ensaios clnicos eram as sulfonamidas e as drogas antimalricas. Colebrook e Perdie mostraram uma reduo na mortalidade de 22 para 8%, com uso de sulfonamidas na febre psparto, comparando com o grupo controle, com o histrico de pacientes tratados no mesmo hospital (COLEBROOK; PURDIE, 1937).
A descoberta da penicilina pode ser considerada o mais importante avano teraputico do sculo vinte, levando elaborao de um ensaio clnico com grande nmero de pacientes e mdicos colaboradores que teve lugar no norte da frica. Aps os primeiros resultados, os mdicos passaram a utiliz-la em pacientes mais graves do que aqueles propostos, quase chegando a prejudicar o resultado final do estudo (BLUMBERG; STROMINGER, 1974).
geralmente aceito que o primeiro ensaio clnico, com um grupo de controle aleatrio (randomizado), foi realizado para estreptomicina, em tratamento de tuberculose pulmonar. Este ensaio foi notvel pelo grau de cuidado com o planejamento, execuo e relato, traos desejados nos ensaios clnicos atuais. Vrios centros foram utilizados, alm da distribuio aleatria para o tratamento. Os grupos foram divididos em tratados com o frmaco e repouso e apenas repouso. Colocadas em envelopes lacrados, as radiografias foram avaliadas por trs mdicos, de forma independente e sem conhecimento prvio de outros laudos e do tratamento aplicado ao paciente. Os resultados validaram amplamente o uso de estreptomicina.
77
Outro estudo, por sua vez, demonstrou que o uso de anti-histamnicos no tratamento da gripe no apresenta vantagens ao ser comparado com o placebo. Neste estudo foi introduzido o duplo cego, onde nem o mdico nem paciente tinham conhecimento prvio da formulao administrada (BULL, 1959).
Sir Austin Bradford Hill foi o principal pesquisador envolvido com ensaios clnicos no Medical Research Council tendo organizado vrios estudos clnicos e publicado diversos artigos sobre conduo destes protocolos, demonstrando a importncia da escolha dos pacientes, randomizao, avaliao objetiva e anlise estatstica (ATKINSON et al., 2001).
Os motivos que retardaram o progresso dos ensaios clnicos foram reverncia autoridade, relacionamento entre mdico e paciente, poucos registros, falta de facilidades para investigao e falta de remdios ativo. A razo mais importante para o desenvolvimento de novos estudos a crescente preocupao com o tratamento de doenas no transmissveis (BULL, 1959).
A indstria farmacutica sofreu uma enorme expanso nos ltimos 40 anos e, com isso, novas drogas foram descobertas e sintetizadas. Antes da II Guerra Mundial no havia qualquer controle para que um frmaco fosse livremente comercializado. Contudo nos Estados Unidos j existia, desde 1938, legislao para uso de frmacos em animais. Foi apenas na dcada de 60, com a catstrofe da talidomida, que tanto os Estados Unidos como a Inglaterra, intensificaram as regulamentaes para a execuo de ensaios em humanos. A partir de 1963 obrigatria uma aprovao oficial para que o frmaco inicie um ensaio clnico e, posteriormente, nova aprovao para ser comercializada no Reino Unido. Nos Estados Unidos, o FDA (Food and Drug Administration) desenvolveu e aplicou diretrizes (a partir da dcada de 70) para a elaborao de um modelo seguro de pesquisa clnica, seguido por muitos outros pases, diferindo do modelo britnico, principalmente por valorizar sobremaneira a documentao dos dados obtidos (ATKINSON et al., 2001).
78
, sem dvida, verdadeira a afirmao de que hoje se faz mais ensaios clnicos do que em outras pocas, sendo em sua grande maioria financiados pela prpria indstria farmacutica. Na ltima dcada tem-se investido em trs linhas de frmacos: as de ao antiinfecciosa, as que atuam no sistema nervoso central e as ativas sobre sistema cardiovascular. Apenas um em dez mil frmacos sintetizados atinge a fase de ensaio clnico, isso explica porque gasto mais com ensaios prclnicos do que com clnicos. Alm disso, apenas cerda de 20% destas geralmente comercializada. Todo este processo gasta cerca de 7 a 10 anos para colocar um novo frmaco no mercado, com um custo estimado de 54 milhes de dlares (ATKINSON et al., 2001).
No Brasil houve um grande salto (tanto qualitativo como quantitativo) nos trabalhos cientficos relacionados farmacologia clinica, com o surgimento da poltica de medicamentos genricos (ANVISA, 2001).
4 ESTUDOS DE BIOEQUIVALNCIA
A biodisponibilidade refere-se quantidade de determinada droga encontrada no sangue, aps sua absoro no local onde foi administrada. Os valores de concentrao no sangue de um frmaco administrado por via oral, quando comparados com os valores do mesmo frmaco administrado por via intravenosa, do-nos o parmetro conhecido como biodisponibilidade absoluta. Quando comparao feita com outro frmaco, chamado de referncia, administrado pela mesma via, chamamos de biodisponibilidade relativa. Diversos fatores relacionados com o frmaco em teste meia-vida, induo enzimtica, sinergismo ou com o paciente idade, sexo, jejum, uso de outros medicamentos podem causar importantes alteraes na biodisponibilidade (ATKINSON et al., 2001; FINK, 1998; FDA, 1985; FDAa, 1988; FDAb, 1988; FDA, 1993; GOODMAN; GILMAN, 2007; PAGE et al., 2002).
Dois medicamentos so considerados equivalentes farmacuticos quando possuem os mesmos integrantes ativos e so idnticos quanto formulao e a via de administrao. Para que possam ser considerados bioequivalentes devem
79
apresentar taxa e extenso de absoro do princpio ativo, bem como a excreo, dentro dos limites aceitos por estatstica adequada. Para tanto, os seguintes parmetros farmacocinticos devem ser levados em considerao (ANVISA, 2001; ATKINSON et al., 2001; FINK, 1998; FDA, 1985; FDAa, 1988; FDAb, 1988; FDA, 1993; GOODMAN; GILMAN, 2007; PAGE et al., 2002):
Cmax Pico de concentrao do frmaco no sangue, referindo-se ao valor mximo atingido pelo frmaco aps sua administrao.
Tmax Tempo no qual ocorre o pico de concentrao do frmaco no sangue aps sua administrao.
AUC rea sob a curva de concentrao numa unidade de tempo. Enquanto Cmax e Tmax so obtidos apenas montando-se o grfico das dosagens do frmaco, a AUC pode ser determinada utilizando-se a regra da soma dos trapzios.
Segundo a resoluo da ANVISA, RDC N 10, de 02 de janeiro de 2001, que estabelece os critrios para provas de bioequivalncia de medicamentos genricos, estas devero contemplar trs etapas: clnica, analtica e estatstica (ANVISA, 2001).
4.1 Etapa Clnica
Os medicamentos a serem submetidos ao estudo de biodisponibilidade devero, inicialmente, ser analisados segundo sua monografia inscrita na Farmacopia Brasileira e, na falta desta, em outros cdigos autorizados pela legislao vigente. O estudo de biodisponibilidade realizado, geralmente, por meio da qualificao do frmaco ou do metablito ativo na circulao (freqentemente em plasma ou soro), ou atravs de sua quantificao na urina, quando justificado (ANVISA, 2001; ATKINSON et al., 2001; FINK, 1998; FDA, 1985; FDAa, 1988; FDAb, 1988; FDA, 1993).
80
O estudo de biodisponibilidade do tipo aberto, aleatrio, cruzado. Os voluntrios recebem os medicamentos teste e referncia (medicamento administrado por via intravenosa ou, quando no for indicada, uma soluo oral do frmaco) em ocasies separadas (perodos), em esquema de dose simples ou mltipla. O intervalo entre os perodos deve ser de, no mnimo, sete meias-vidas de eliminao do frmaco, ou do metablito, quando o mesmo for ativo (ANVISA, 2001; ATKINSON et al., 2001; FINK, 1998; FDA, 1985; FDAa, 1988; FDAb, 1988; FDA, 1993).
O cronograma de coleta das amostras deve contemplar um tempo igual ou superior 3-5 vezes a meia-vida de eliminao do frmaco, ou do metablito, quando o mesmo for ativo e o nmero mnimo de voluntrios sadios dever ser de 24, com idade entre 18 e 50 anos e capazes de fornecer seu consentimento livre e esclarecido. Os estudos podem ser conduzidos com voluntrios do sexo masculino, feminino ou ambos, sendo que neste caso, o nmero de homens e mulheres deve ser o mesmo. A ANVISA pode exigir um nmero maior de voluntrios para frmacos que apresentem grande variabilidade (ANVISA, 2001).
O peso dos voluntrios deve estar em um limite de +/- 15% do peso considerado normal, levando-se em considerao a altura e estrutura fsica. Devemse evitar indivduos fumantes e com histrico de abuso de lcool ou drogas. Caso sejam includos fumantes, os mesmos devem estar identificados (ANVISA, 2001; ATKINSON et al., 2001; FINK, 1998; FDA, 1985; FDAa, 1988; FDAb, 1988; FDA, 1993).
O projeto de pesquisa, o protocolo experimental e o termo de consentimento livre e esclarecido devem ser submetidos a um Comit de tica em Pesquisa (CEP), credenciado no Comit Nacional de tica em Pesquisa (CONEP) do Conselho Nacional de Sade/MS (ANVISA, 2001).
Os voluntrios participantes dos estudos clnicos, que necessitem de confinamento, devem permanecer em local apropriado que atenda s Boas Prticas
81
Clnicas (BPC), sob a responsabilidade de um mdico (ANVISA, 2001; ATKINSON et al., 2001; FINK, 1998; FDA, 1985; FDAa, 1988; FDAb, 1988; FDA, 1993).
4.2 Etapa Analtica
Todas as etapas do estudo devem ser realizadas de acordo com as normas internacionais de Boas Prticas de Laboratrio (BPL) e os mtodos analticos devem ser validados. O protocolo analtico deve conter os critrios para reanlise das amostras. No mais do que 20% das amostras podem ser reanalisadas e a perda de amostras em qualquer etapa do processo analtico deve ser justificada (CAUSON, 1997).
A anlise das amostras pode ser efetuada nas seguintes condies: sem rplica, em duplicata ou triplicata. Para anlise de amostras em duplicata, deve-se utilizar o valor mdio, e para triplicata, a mdia dos dois valores mais prximos (ANVISA, 2001).
A determinao da adequabilidade e confiabilidade de um mtodo analtico para a realizao de um estudo de bioequivalncia realizada atravs da averiguao de sua sensibilidade, especificidade, linearidade, exatido, preciso e reprodutibilidade, levando-se ainda em conta a estabilidade dos compostos que estejam sendo empregados (ANVISA, 2001; ATKINSON et al., 2001; FINK, 1998; FDA, 1985; FDAa, 1988; FDAb, 1988; FDA, 1993).
O quadro 3 apresenta os parmetros utilizados para a validao do mtodo desenvolvido (CHASIN et al., 1994).
82
Quadro 3 Parmetros utilizados na validao do mtodo.
Parmetro
Recuperao Linearidade Repetibilidade
Definio
Eficincia do processo de extrao Faixa de concentrao plasmtica com relao linear entre concentrao e resposta Preciso intra-ensaio: variao dos resultados em anlises realizadas no mesmo dia. Preciso interensaio: variao dos resultados em anlises realizadas em dias consecutivos.
Unidade
% ng/mL ou ug/mL CV%
Exatido LQ Seletividade
Avalia a variao entre o valor real e o valor obtido experimentalmente Limite de quantificao: menor concentrao do frmaco quantificado com erro inferior a 20% Interferentes endgenos
% ng/mL ou ug/mL min
* C.V. = coeficiente de variao.
4.3 Etapa Estatstica e Farmacocintica
Os parmetros farmacocinticos so obtidos das curvas de concentrao sangunea do frmaco versus tempo e analisados, estatisticamente, para determinao da biodisponibilidade. Os seguintes parmetros farmacocinticos devem ser determinados (ANVISA, 2001; ATKINSON et al., 2001; FINK, 1998; FDA, 1985; FDAa, 1988; FDAb, 1988; FDA, 1993): - rea sob a curva de concentrao sangunea versus tempo, calculada pelo mtodo dos trapezides, do tempo zero ao tempo t (AUC0-t), onde t o tempo relativo ltima concentrao determinada experimentalmente. - rea sob a curva de concentrao sangunea versus tempo, calculada do tempo zero ao tempo infinito (AUC0-inf), onde AUC0-inf = AUC0-t + Ct/Ke, onde Ct a ltima concentrao do frmaco determinada experimentalmente e Ke a constante de eliminao da fase terminal. A AUC0-t deve ser igual ou superior a 80% da AUC0-inf. - O pico de concentrao mxima (Cmax) do frmaco e/ou metablito e o tempo de atingir este pico (Tmax) devem ser obtidos diretamente sem interpolao dos dados.
83
- A depurao (D), o volume aparente de distribuio (Vd) e a meia-vida de eliminao (t1/2) do frmaco e/ou metablito tambm devem ser determinados, embora no haja necessidade de tratamento estatstico. - A biodisponibilidade absoluta (F) do medicamento deve ser determinada e correspondente frao da dose administrada do frmaco efetivamente absorvida.
5 MEDICAMENTOS GENRICOS
Os genricos so medicamentos que possuem o mesmo princpio ativo, na mesma dose e na mesma forma farmacutica, sendo administrados pela mesma via e com a mesma indicao teraputica do medicamento de referncia, com o qual devem ser intercambiveis. A adoo de uma poltica de medicamentos genricos, envolvendo a produo, a garantia de qualidade, a prescrio, a dispensao e o uso dos mesmos, parte fundamental de uma diretriz para a promoo e o uso racional de medicamentos em nosso Pas. A poltica de medicamentos genricos objetiva maior racionalidade no uso de medicamentos, bem como estimula a concorrncia, na qual os consumidores tero disponveis produtos intercambiveis de diferentes preos. previsvel que a referida competio ocasione a reduo dos preos dos medicamentos, trazendo, ento, benefcios a todos os segmentos envolvidos na cadeia de produo, controle, comercializao e, principalmente, o consumo. A adoo de poltica de medicamentos genricos no Brasil objetivou principalmente aumentar o acesso da populao aos medicamentos, melhorar a qualidade dos mesmos e diminuir o custo dos tratamentos mdicos.
No Brasil, a questo dos genricos comea a tomar flego, a partir da Lei 9787, sancionada pelo presidente da Repblica Fernando Henrique Cardoso, em 10 de fevereiro de 1999, que estabelece o que medicamento genrico e dispe sobre a utilizao de nomes genricos em produtos farmacuticos, sob fiscalizao da ANVISA (Agncia Nacional de Vigilncia Sanitria). Essa legislao foi revista e atualizada com o objetivo de melhor esclarecer as exigncias, em relao aos medicamentos genricos atravs da RDC 210 de janeiro de 2001. Posteriormente, foi editada a RDC 84, de maro de 2002, com o intuito de detalhar as regulamentaes j previstas na RDC 10. Esta nova resoluo transforma os anexos
84
da RDC 10 em Guias publicados na forma de resolues (RE), como por exemplo, a RE 478-Guia para provas de bioequivalncia de medicamentos genricos (RIBEIRO, 2000).
O surgimento dos produtos denominados genricos aparece como uma alternativa aos produtos tradicionais e podem revolucionar as questes inerentes falta de recursos dos governos para subsidiar a populao de baixa renda (ABIFAM, 2001).
CAPTULO 4: Medicamentos Avaliados
1 AMOXICILINA
A penicilina o prottipo dos antibiticos betalactmicos. o agente antimicrobiano de escolha para o tratamento e profilaxia de varias enfermidades infecciosas. A sensibilidade de um microorganismo aos antibiticos betalactmicos depende de sua capacidade de penetrar na parede bacteriana e ligar-se em sua forma ativa ao local visado. O mecanismo de ao destes antibiticos ainda no totalmente conhecido, mas acredita-se que eles so capazes de interferir na sntese da parede bacteriana dos organismos em atividade (TOMASZ, 1979a; TOMASZ, 1979b).
Aps
penetrar
na
bactria,
antibitico
betalactmico
inativa
transpeptidases, carboxipeptidases e endopeptidases, interferindo na sntese da parede celular (BLUMBERG; STROMINGER, 1974; WAXMAN; STROMINGER, 1983). A amoxicilina (alfa-amino-p-hidrobenzilpenicilina) uma penicilina semisinttica estruturalmente relacionada com a ampicilina que apresenta atividade oral, sendo que seu espectro de atividade semelhante ao da ampicilina. Seu ncleo consiste de um anel tiazolidnico, ligado a um anel B-lactmico com uma cadeia lateral. Esta cadeia lateral determina a maior parte da atividade farmacolgica e antimicrobiana da penicilina em questo. No caso da amoxicilina, a presena do anel benzil na cadeia lateral estende sua atividade antimicrobiana a bactrias gramnegativas. Pode ser utilizada por via oral e parenteral (sal sdico-C16H18N3NaO5S)
85
(GORDON et al., 1972; GORDON; WINSHELL, 1970; NEU; WINSHELL, 1970; SUTHERLAND; ROLINSON, 1970). A amoxicilina apresenta elevada absoro aps administrao oral, sendo que esta no alterada pela ingesto concomitante a alimentos. Mostra baixa ligao com protenas plasmticas (17%) e rapidamente distribuda pelo corpo, exibindo uma meia-vida de eliminao de 1h. A eliminao da droga se d preferencialmente pelos rins, com cerca de 60% da dose administrada por via oral e 75% da parenteral sendo excretada inalterada (BARR et al., 1994; GORDON et al., 1972; GORDON; WINSHELL, 1970; HANDSFIELD et al., 1973; KOSMIDIS et al., 1972; NEU; WINSHELL, 1970; NEU, 1974; PAINTAUD et al., 1992; SIMON; STILE, 1985).
As concentraes plasmticas mximas da amoxicilina so duas a duas vezes e meia maiores que as da ampicilina aps administrao oral da mesma dose. Essas concentraes so obtidas em duas horas e atingem, em mdia cerca de 4 g/mL quando se administram 250 mg. Apesar da meia-vida semelhante da amoxicilina e da ampicilina, as concentraes plasmticas eficazes da amoxicilina administrada por via oral so detectveis por um perodo duas vezes maior do que quando se administrada a ampicilina, por causa da sua absoro mais completa (GOODMAN; GILMAN, 2007).
Os mtodos mais utilizados para a quantificao da amoxicilina em fludos biolgicos empregam a cromatografia lquida de alta eficincia (CLAE), sendo que na maior parte destes mtodos aplicada a deteco no UV em baixos comprimentos de onda, = 225 330 nm (MUTH et al., 1996; HOIZEY et al., 2002; SOURGENS et al., 2001; YUAN et al., 1995; KRAUWINKEL et al., 1993; CHARLES; CHULAVATNATOL, 1993). O LQ mais baixo obtido por essas metodologias foi 50 ng/mL. Contudo, os tempos de eluio da amoxicilina e do padro interno, cefadroxil, foram 31,8 e 32,8 min, respectivamente, o que dificulta sua aplicao na anlise de rotina (MUTH et al., 1996). A formao de um derivado para a deteco por fluorimetria (MARSCHER; KIKUTA, 1990; WIBAWA et al., 2002; MIYAZAKI et al., 1983), reagentes de formao de par inico e tcnicas especiais de deteco como derivao ps-coluna ou acoplamento de colunas (MUTH et al., 1996; MARSCHER & KIKUTA, 1990) tambm tem sido empregadas para aumentar a
86
sensibilidade e a seletividade, porm so procedimentos caros, trabalhosos e que consomem muito tempo.
Diferentes mtodos de extrao da amostra tm sido aplicados antes da anlise cromatogrfica, sendo que a maioria baseada na precipitao de protenas (HOIZEY et al., 2002; CHARLES; CHULAVATNATOL, 1993; MARSCHER; KIKUTA, 1990), extrao lquido-lquido (WIBAWA et al., 2002) ou procedimentos mais complicados como a extrao em fase slida (YUAN et al., 1995; KRAUWINKEL et al., 1993; MAURER, 1998; HENION et al., 1998). Dentre estas abordagens a extrao por precipitao ser selecionada por ser mais simples, rpida e barata.
A amoxicilina atualmente o antibitico mais utilizado a nvel mundial. Para a determinao dos parmetros farmacocinticos da amoxicilina em humanos necessrio utilizar um mtodo quantitativo confivel.
2 NORFLOXACINO
Norfloxacino
um
frmaco
antimicrobiano
da
classe
das
fluoquinolonas, de estrutura qumica: C16H16FN3O3 - cido-1-etil-6-fluor-1,4dihidro-4-oxo-7-(1piperazinil)-3-quinolinico-carboxlico. O norfloxacino preparado por sntese qumica. O composto um p branco ou amarelo fraco, cristalino e desprovido de odor e com um sabor amargo.
O norfloxacino aparentemente, assim como outras quinolonas, inibe a sntese e/ou conformao do DNA atravs da inibio da DNA girase (topoisomerase II), uma enzima responsvel pelo enovelamento negativo, dependente de ATP do DNA bacteriano (COZZARELLI, 1990, WOLFSON; HOOPER, 1985, BENBROOK; MILER, 1986, HOOPER; WOLFSON, 1989). Alguns investigadores acreditam que o frmaco age produzindo um anexo covalente da DNA girase diretamente no DNA, produzindo um complexo que inacessvel a DNA polimerase (ENGLE et al., 1983). O efeito bactericida do norfloxacino antagonizado pela rifampicina e o cloranfenicol e parece envolver a interrupo da
87
sntese protica. O mecanismo responsvel pela ao letal sobre a bactria no est completamente esclarecido (CRUMPLIN et al., 1984).
As quinolonas podem ser conjugadas com o cido glucornico na posio 3, mas esta o metablito menor do norfloxacino. Os metablitos principais so derivados da substituio do anel piperaznico. Estes ocorrem pela transformao do amino-nitrognio e com a formao de derivados formil ou acetil, ou pela oxidao do tomo de carbono no anel piperaznico em um grupo ceto designado como oxoderivativo. Os metablitos do anel aberto so formados pela hidroxilao ou carboxilao do anel piperaznico, levando formao de compostos acetilaminoetil, desentil e amino do metabolismo progressivo da estrutura do anel.
A oxo-piperazina o metablito mais encontrado na urina, perfazendo 5% do total da dose. Os derivados acetil e formil piperaznicos do norfloxacino perfazem apenas 0,5% do total excretado na urina. Entre os metablitos do anel aberto, o disentil perfaz mais do que os metablitos acetilamino e amino combinados. Metade da dose do norfloxacino recuperada na urina, com compostos e metablitos similares, e a outra metade excretada de forma extra-renal. Os metablitos excretados de forma extra-renal so quantificados de forma inadequada, mas sugere-se que a proporo do frmaco metabolizado maior do que a observada na urina. Quadro 4 Parmetros Farmacocinticos do Norfloxacino. Absoro oral Metabolismo pr-sistmico Meia-vida plasmtica (faixa) Volume de distribuio Ligao protica plasmtica 50 - 80% no observado 3 horas 2,5-3,1 kg-1 15%
Aps uma dose oral de 200 ou 400 mg o pico das concentraes sricas alcanadas em 60 a 90 minutos so de 0,8 0,3 e 1,5 0,6 mg.L-1, respectivamente. A absoro levemente alterada quando o norfloxacino administrado juntamente com a comida, com uma reduo de 30% no pico de
88
concentrao. A administrao de 400 mg a cada 12 horas provoca uma pequena acumulao, sem alterao nas concentraes sricas.
H pouca informao sobre a penetrao do norfloxacino em diferentes tecidos. Aps a administrao oral de norfloxacino, a concentrao nos tecidos vaginais, cervical, nas trompas, nos ovrios, no crtex renal e na vescula biliar foi discretamente menor que a concentrao srica. A blis, o fgado e a medula da adrenal obtiveram concentraes de droga mais altas que as sricas. A concentrao do escarro foi relativamente baixa. O norfloxacino no foi encontrado no leite humano aps dose oral de 200 mg. Aps a ingesto de 400 mg de norfloxacino, o pico na urina foi de 300 mg.L-1, o que centenas de vezes mais alta do que a CIM da maioria das bactrias patognicas que causam infeces entricas.
O norfloxacino excretado na urina atravs de excreo. A excreo renal do norfloxacino ocorre atravs da filtrao glomerular e da secreo tubular. O clearance renal de 275 71mL/min. O probenecid reduz a excreo renal em 50% (NORRBY, 1983; WISE, 1984; BERGERON et al., 1984; COFSKY et al., 1984; SWANSON et al., 1983).
A meia-vida de eliminao de 3 horas (2,5 - 4,5h). Dentro de 24 horas da administrao de 26 a 32% eliminado na urina sob a forma de norfloxacino e de 5 a 8% como metablitos adicionais. Captao fecal perfaz 30% da dose administrada e 2 a 3% eliminada pela blis.
O mtodo preferido para anlise o da cromatografia lquida de alta eficincia (HPLC). A HPLC com deteco de fluorescncia recomendada, uma vez que rpida, sensvel e especfica. Os metablitos tambm podem ser detectados pelo HPLC. As limitaes na sensibilidade de detectar compostos similares e metablitos variam de acordo com o mtodo e o equipamento.
89
3 OXCARBAZEPINA
A oxcarbazepina um anlogo da carbamazepina. um derivado iminodibenzlicos com um grupo carbamila na posio 5, sendo estruturalmente diferente nas posies 10 e 11. rapidamente convertida em 10-hidroxi-10,11dihidrocarbazepina (monohidroxi derivado - MHD), o qual responsvel por grande parte de sua atividade farmacolgica. A oxcarbazepina possui atividade anticonvulsivante, apresentando estrutura qumica semelhante aos antidepressivos tricclicos (ANDERSON; LEVY, 1995; BIALER, 2002; MCLEAN et al., 1994).
A oxcarbazepina e seu metablito farmacologicamente ativo MHD apresentam efeito antiepilptico potente quando comparada em modelos animais com a carbamazepina e fenitona. Acredita-se que o mecanismo de ao da oxcarbazepina e MHD seja baseado principalmente no bloqueio de canais do sdio voltagem-dependentes, resultando ento na estabilizao de membranas neurais hiperexcitadas, inibio da descarga neuronal repetitiva e diminuio da propagao de impulsos sinpticos. Adicionalmente, o aumento na condutncia de potssio e modulao de canais de clcio voltagem-dependentes ativados pode tambm contribuir para os efeitos anticonvulsivantes das drogas (GLAUSER, 2001; GRANT; FAULDS, 1992; HOUTKOOPER et al., 1987; LLOYD et al., 1994; MCLEAN et al., 1994). Quadro 5 Parmetros Farmacocinticos do Oxcarbazepina e MHD. Parmetros
Absoro oral Metabolismo heptico Meia-vida plasmtica (faixa) Tmax Volume de distribuio Clearance Ligao protica plasmtica 50-70%
Oxcarbazepina
~100% Extenso 1,3 2,3 h 1-2 horas Conflituoso
MHD
--9,3 1,8h 4-6 horas 0,3-0,8 L/kg 13,6 20,8 mL/min 35-50%
90
A oxcarbazepina completamente absorvida. A alimentao no tem nenhum efeito sobre a absoro desta droga, podendo ser administrado com ou sem alimentao. Os picos de concentrao plasmtica da droga e de seu metablito so obtidos num perodo de 1-2 horas e 4-6 horas, respectivamente. Aps a administrao oral da oxcarbazepina marcada radioativamente, apenas 2% da radioatividade total no plasma devida molcula inalterada da oxcarbazepina e aproximadamente 70% devida ao metablito MHD (MAY et al., 2003; LLOYD et al., 1994).
Dados sobre o volume de distribuio da oxcarbazepina so conflituosos. Hopper (1987) relatou um volume aparente mdio desta droga de 3,9 L/Kg, enquanto Dickinson e colaboradores calcularam uma mdia aparente do volume de distribuio de 7,8 a 12,5 L/Kg.
A oxcarbazepina rapidamente transformada no seu metablito ativo (MHD) por enzimas citoslicas no fgado. O MHD metabolizado pelo cido glicurnico. Quantidades menores (4% da dose) so convertidas ao metablito farmacologicamente inativo da droga (KRISTENSEN et al., 1983; SCHIITZ et al., 1986).
A oxcarbazepina eliminada do organismo principalmente sob formas de metablitos, os quais so principalmente excretados pelos rins. Mais de 95% da dose aparece na urina, com menos de 1% como oxcarbazepina inalterada. A quantidade eliminada nas fezes representa menos de 4% da dose administrada (ROUAN et al., 1994).
A oxcarbazepina rapidamente eliminada do plasma com valor aparente de meia-vida plasmtica entre 1,3 e 2,3 horas. A meia-vida plasmtica aparente do MHD 9,3 1,8h. Quando a oxcarbazepina administrada duas vezes ao dia, as concentraes plasmticas de estado de equilbrio so alcanadas dentro de 2-3 dias. Em virtude de sua meia-vida curta, a oxcarbazepina rapidamente eliminada do plasma e apenas baixas concentraes de oxcarbazepina resultam em dosagens teraputicas. Valores publicados do clearance renal de MHD permaneceram no
91
intervalo entre 13,6 a 20,8 mL/min (NEDELMAN al.,1990; PATSALOS et al., 2003).
et al., 1999; PATSALOS et
De acordo com a literatura, os primeiros procedimentos utilizavam a cromatografia gasosa para determinao da oxcarbazepina e MHD no soro ou no plasma. Atualmente, pesquisadores tm se baseado em sistemas de cromatografia lquida de alta eficincia. Alm disso, a cromatografia gasosa acoplada espectrometria de massa tambm vem sendo desenvolvida (VON UNRUH; PAAR, 1985; VON UNRUH; PAAR, 1986; MENGE; DUBOIS, 1983, MENGE et al., 1987; MATAR et al., 1995; LEVERT et al., 2002; JUERGENS, 1987).
CAPTULO 5: Justificativa
A confiabilidade dos medicamentos pode ser assegurada por meio de rgidos critrios de qualidade exigidos para a anlise na concesso do seu registro, previstos na legislao. A garantia desses padres de qualidade constitui uma ferramenta para a segurana, eficcia e qualidade dos medicamentos.
A fim de alcanar a qualidade dos medicamentos, alm dos processos de manufatura, observando as boas prticas de fabricao, fundamental a realizao de estudos de farmacocintica comparada de frmacos, para verificar existncia de bioequivalncia entre o medicamento teste e o medicamento referncia adotado como padro pela Agncia Nacional de Vigilncia Sanitria Anvisa. Tambm se torna necessrio o desenvolvimento de metodologias de baixo custo e fcil aplicao, com sensibilidade e seletividade adequadas para realizao deste tipo de estudo. Nesse contexto, cresce a necessidade e a importncia do desenvolvimento e validao de metodologias para quantificao dos frmacos amoxicilina, norfloxacino, oxcarbazepina e 10,11-dihidro-10-hidroxicarbamazepina, em plasma, destacando a gerao de dados reprodutveis e confiveis aps execuo de estudos farmacocinticos, uma vez que so medicamentos utilizados no Sistema nico de Sade (SUS).
92
PARTE II: OBJETIVOS E METODOLOGIA
93
PARTE II: OBJETIVOS E METODOLOGIA
CAPTULO 1: Objetivos
1 OBJETIVO GERAL
Desenvolver e validar uma metodologia para quantificao de amoxicilina, norfloxacino, oxcarbazepina e seu metablito, em plasma humano, por cromatografia lquida de alta eficincia acoplada ao detector ultravioleta, fluorescncia ou espectrometria de massa, respectivamente.
2 OBJETIVOS ESPECFICOS
Determinar o perfil farmacocintico e a bioequivalncia de formas farmacuticas orais contendo amoxicilina, norfloxacino, oxcarbazepina e seu metablito em voluntrios sadios.
Aplicar metodologia analtica para quantificao das formulaes de amoxicilina, cpsulas de 500mg, do laboratrio Glenmark Pharmaceuticals Ltda (teste) e laboratrio GlaxoSmithKline Beecham (formulao referncia) em estudo de bioequivalncia.
Aplicar metodologia analtica para quantificao das formulaes de norfloxacino, comprimido de 400mg, do laboratrio Pharmascience Laboratrios Ltda (formulao teste) e laboratrio Merck Sharp & Dohme Farmacutica Ltda (formulao referncia) em estudo de bioequivalncia.
Aplicar metodologia analtica para quantificao das formulaes de oxcarbazepina e seu metablito, comprimido de 400mg, do laboratrio Novartis Biocincias S.A. na determinao do perfil farmacocintico.
94
CAPTULO 2: Metodologia
1 PROTOCOLO CLNICO
1.1 Local de Confinamento dos Voluntrios
Os voluntrios foram confinados na Unidade de Farmacologia Clnica do Departamento de Fisiologia e Farmacologia da Faculdade de Medicina da Universidade Federal do Cear. Esta unidade de ensaios clnicos dispe de uma estrutura assistencial prpria, que consiste de uma unidade para ensaios clnicos com at 24 leitos e posto de enfermagem.
A Unidade de Farmacologia Clnica (UNIFAC) dispe de carrinho de emergncia com desfibrilador e monitor cardaco, oxmetro, respirador, material para pequena cirurgia e com medicao de urgncia para qualquer eventualidade. Alm disso, conta com apoio de uma Unidade de Terapia Intensiva (UTI) do Hospital de Messejana. Sua estrutura laboratorial possui equipamentos para processamento e armazenamento de amostras biolgicas.
1.2 Delineamento do estudo
A pesquisa consistiu de trs estudos abertos, randomizados, com replicao para 48 voluntrios sadios, includos para o estudo da Amoxicilina e Norfloxacino, e uma administrao simples de Oxcarbazepina para oito voluntrios sadios, sendo todos adultos, de ambos os sexos e com idade de 18 a 40 anos.
Depois da seleo e observado um perodo de pelo menos 4 semanas sem fazer uso de qualquer medicao, os voluntrios qualificados para participar do estudo foram internados por dois perodos variando de 24 a 36 horas, com intervalo de 7 dias entre os internamentos (wash-out), afim de se evitar efeitos residuais (carry-over). Os voluntrios apresentaram-se para internamento na UNIFAC, entre 20:00 e 22:00 horas da noite anterior administrao da medicao. Em cada
95
internamento, os voluntrios receberam as formulaes contendo Amoxicilina, Amoxil, Norfloxacino, Floxacin e Oxcarbazepina.
1.3 Seleo dos voluntrios
A avaliao clnica inicial (pr-estudo) consistiu em uma histria clnica a fim de pesquisar sintomas relativos a alergias, manifestaes relativas aos olhos, nariz, ouvidos, orofaringe, sistema respiratrio, cardiovascular, gastrintestinal, geniturinrio, sistema nervoso central, linfo-hematopotico, endcrino e metablico, musculoesqueltico e dermatolgico. Aspectos relativos estabilidade emocional, antecedente de hipersensibilidade drogas, antecedentes cirrgicos, antecedentes patolgicos familiares e outros, considerados importantes no momento, tambm foram investigados.
No exame fsico foram registrados: a altura (em centmetros), o peso corporal (em kilogramas), com posterior clculo do ndice de Massa Corporal (IMC), e a temperatura axilar (em graus Celsius). Exame dos segmentos corporal foi efetuado, avaliando-se o aspecto geral, a pele, cabea e pescoo, olhos, ouvidos, nariz, boca, orofaringe, trax e pulmes, corao, abdmen, coluna vertebral, genitlia, linfonodos, extremidades.
1.3.1 Sinais vitais
No perodo de realizao do pr-estudo, ps-estudo e durante o estudo clnico a presso arterial diastlica e sistlica, a freqncia de pulso e a temperatura axilar foram aferidas e registradas no Formulrio de Relato de Caso (CRF).
1.3.2 Exame cardiolgico
Eletrocardiograma
convencional
com
12
derivaes
foi
realizado
juntamente com os outros exames laboratoriais antes e aps o final da etapa clnica. Os parmetros: SP (vetor resultante da despolarizao atrial), SQRS (vetor resultante da despolarizao ventricular), ST (vetor resultante da repolarizao
96
ventricular), ritmo e freqncia cardaca, durao do complexo QRS e dos intervalos P-R e Q-T, foram registrados.
Foram observadas tambm a presena de distrbios da conduo intraventricular e atrioventricular, sinais de sobrecargas de cmaras atriais e ventriculares. O laudo final era referido como normal, anormal no clinicamente significante ou anormal com significado clnico.
1.3.3 Exames laboratoriais
Os seguintes exames laboratoriais foram realizados nos perodos prestudo (perodo de avaliao para seleo de voluntrios saudveis) e ps-estudo (perodo aps o segundo e ltimo internamento).
a) Anlise Hematolgica: Hemoglobina, Hematcrito, Contagem total e diferencial de leuccitos, Contagem de plaquetas e Velocidade de Hemossedimentao (VHS).
b) Anlise bioqumica: Uria, Creatinina, Bilirrubina Total, Protena Total, Albumina, Glicose em jejum, Fosfatase alcalina, Aspartato-amino-transferase (AST), Alanina-amino-transferase (ALT), Colesterol total, Triglicrides, cido rico e Gama GT.
c) Sumrio de urina
d) Anlise sorolgica: Hepatite B, Hepatite C, HIV (Sndrome da Imunodeficincia Adquirida - AIDS) e teste sorolgico para gravidez (Beta-HCG) para voluntrios do sexo feminino.
Exame realizado somente no pr-estudo: Anlise Sorolgica.
97
1.3.4 Valores de referncia dos exames laboratoriais
Os valores utilizados como referncia para os exames laboratoriais foram fornecidos pelo laboratrio de patologia clnica responsvel por estas anlises, Laboratrio Louis Pasteur .
Os parmetros de normalidade para a anlise sorolgica para hepatite B, C e HIV devem consistir de resultados negativos.
1.4 Critrios de incluso
Os seguintes critrios devem ser satisfeitos para que o voluntrio possa participar do estudo:
a) Homem ou Mulher com idade entre 18 a 50 anos. As mulheres no poderiam estar grvidas e nem em regime de amamentao.
b) Voluntrio com ndice de massa corprea maior do que 19 e menor do que 28.
c) Boas condies de sade ou sem doenas significativas, a juzo mdico, de acordo com as regras definidas no protocolo, e avaliaes a que foi submetido: histria clnica, medidas de presso e pulso, exame fsico e psicolgico, ECG e exames laboratoriais complementares.
d) Capaz de compreender a natureza e objetivo do estudo, inclusive os riscos e efeitos adversos e com inteno de cooperar com o pesquisador e agir de acordo com os requerimentos de todo o ensaio, o que vem a ser confirmado mediante a assinatura do Termo de Consentimento Livre e Esclarecido.
98
1.5 Critrios de excluso
A resposta positiva a qualquer um dos seguintes critrios excluir o voluntrio do estudo:
a) O voluntrio tem sabidamente uma hipersensibilidade ao frmaco estudado ou a compostos quimicamente relacionados; histria de reaes adversas srias ou hipersensibilidade a qualquer frmaco.
b) Histria ou presena de doenas hepticas ou gastrointestinais ou outra condio que interfere com a absoro, distribuio, excreo ou metabolismo do frmaco.
c) Uso de terapia de manuteno com qualquer droga, excetuando-se anticoncepcionais por via oral.
d) Tem histria de doena heptica, renal, pulmonar, gastrintestinal, epilptica, hematolgica ou psiquitrica; tem hipo ou hipertenso de qualquer etiologia que necessite de tratamento farmacolgico; tem histria ou teve infarto do miocrdio, angina e/ou insuficincia cardaca;
e) Achados eletrocardiogrficos no recomendados, a critrio do investigador, para participao no estudo.
f) Os resultados dos exames laboratoriais complementares esto fora dos valores considerados normais, de acordo com as normas deste protocolo, a menos que sejam considerados no clinicamente significativos pelo investigador.
g) Voluntrio fumante;
h) O voluntrio ingere mais do que 5 xcaras de caf ou ch por dia;
99
i) Tem histria de abuso de lcool ou drogas;
j) Fez uso de medicao regular dentro das 2 semanas que antecederam o incio deste estudo, ou fez uso de qualquer medicao uma semana antes do incio deste estudo;
l) Foi internado por qualquer motivo at 8 semanas antes do incio do primeiro perodo de tratamento deste estudo;
m) Tratamento dentro dos 3 meses prvios ao incio do tratamento deste estudo com qualquer droga que se conhea ter um potencial txico bem definido nos grandes rgos.
n) O voluntrio participou de qualquer estudo experimental ou ingeriu qualquer droga experimental dentro dos trs meses que antecedem o incio do tratamento deste estudo;
o) O voluntrio doou ou perdeu 450 mL ou mais de sangue dentre dos trs meses que antecederam o estudo ou que doou mais de 1500 mL dentro dos 12 meses anteriores ao incio do tratamento deste estudo.
p) Teste positivo de gravidez para as voluntrias mulheres.
q) O voluntrio tem qualquer condio que o impede de participar do estudo pelo julgamento do investigador
1.6 Critrios para retirada do estudo
a) Solicitao, por parte do voluntrio, para se retirar do estudo a qualquer momento
100
b) Voluntrio no deseja continuar no estudo por razes pessoais (ou mesmo sem relatar a razo);
c) Voluntrio no deseja continuar no estudo devido os eventos adversos da droga de estudo (efeitos no desejveis possivelmente relacionados ao frmaco em estudo)
d) Voluntrio no deseja continuar por razes outras que no efeitos adversos por exemplo indisponibilidade, intolerncia aos procedimentos do estudo.
e) No aderncia s exigncias do protocolo.
f) Eventos adversos ou sintomas ou sinais de possvel toxicidade
g) Doena intercorrente requerendo medicao.
h) Resposta positiva reavaliao de qualquer um dos critrios de excluso, no momento da admisso ao primeiro perodo de tratamento ou em ocasio subseqente.
i) Qualquer outra condio que, a juzo do investigador, seja do interesse para manuteno da sade do voluntrio.
1.7 Entrada do voluntrio no estudo
Os voluntrios foram aceitos no estudo somente quando considerados saudveis como determinado pela histria mdica, exame fsico e exames laboratoriais que antecederam o incio do estudo.
Uma vez avaliada a higidez, os voluntrios foram submetidos a uma entrevista para avaliao das condies emocionais para participar da investigao. Aps todas as dvidas terem sido esclarecidas aos voluntrios, e os mesmos tendo
101
concordado com o protocolo clnico, foi assinado o Termo de Consentimento Livre e Esclarecido (Anexo II) para participao no estudo.
1.8 Restries
Todos os voluntrios chegaram Unidade de Farmacologia Clnica tendo feito uma refeio normal (jantar). A partir das 21:00 horas, da noite do dia do internamento, os voluntrios ficaram em jejum at a manh seguinte e durante o perodo de 3 horas aps a administrao da medicao quando um desjejum foi servido. O almoo foi servido entre 5 e 6 horas aps a administrao e o jantar aps 12 horas. No foram permitidos outros alimentos no perodo de internao. Nos dois perodos os voluntrios tiveram as refeies padronizadas, e lquidos ad libitum aps as refeies, mas bebidas contendo xantinas (incluindo ch, caf e cola) foram evitados.
No foi permitido fumar durante o perodo do internamento. Medicaes concomitantes foram evitadas quando possvel, mas em caso de necessidade, foram registradas em formulrio apropriado. O consumo de lcool foi limitado durante o perodo de estudo e evitado completamente durante as 48 horas que antecederam cada coleta de sangue.
1.9 Esquema experimental
Como relatado anteriormente, os estudos foram abertos e randomizados, sem procedimentos de codificao cega. A seqncia de tratamento, atribuda a cada voluntrio nos perodos de estudo, foi determinada por uma lista de randomizao. O nmero apropriado do voluntrio foi alocado seqencialmente, considerando-se, inicialmente, a ordem alfabtica dos nomes do grupo de voluntrios, cuja entrada na parte randomizada do ensaio for confirmada.
Os voluntrios receberam, conforme a randomizao (ANEXO A e B), um comprimido de amoxicilina, norfloxacino ou suspenso de oxcarbazepina como dose
102
nica entre 7:00 e 8:00 da manh do dia aps o confinamento, acompanhado de 200 mL de gua mineral sem gs, aps pelo menos 10 horas de jejum.
1.10 Coletas de sangue
Retirada de 8 mL de sangue
Centrifugao
Separao 30 L de heparina
Retirada do plasma
Armazenamento
Freezer (-20C)
Figura 11. Esquema do procedimento de coleta de sangue e separao de amostras plasmticas.
As amostras de sangue para determinao da concentrao plasmtica de amoxicilina, norfloxacino, oxcarbazepina e seu metablito foram obtidas, atravs de escalpe heparinizado introduzido em veia superficial do antebrao do voluntrio, imediatamente antes (tempo zero e pool) e aos seguintes intervalos, a partir da administrao: 20, 40, 60, 90, 120, 150, 180, 240, 360, 480, 600; 20, 40, 60, 80, 100, 120, 150, 180, 240, 360, 480, 600, 720, 1080, 1440 e 30, 60, 90, 120, 150, 180, 210, 240, 270, 300, 330, 360, 390, 420, 450, 480, 540, 600, 720, 1080, 1440 e 2880 horas, respectivamente.
A cada intervalo de tempo, foram coletados 8 mL de sangue (exceto antes da administrao quando foram coletados dois tubos de 8 mL cada tempo zero e pool) e colocados em tubos vacutainer de vidro contendo 30 L de heparina. Aps
103
centrifugadas a 3.000 rpm, na temperatura de 8C, durante 12 minutos, o plasma foi separado, colocados em tubos e em seguida os tubos devidamente identificados foram mantidos em freezer, numa temperatura de 20C, at sua anlise (Figura 11).
1.11 Avaliao da segurana
Foi solicitado aos voluntrios que relatassem qualquer evento adverso e a sua data de ocorrncia, para que fosse acompanhado clnica e laboratorialmente at que os parmetros alterados voltassem ao normal. Foi tambm solicitada informao se houve ou no a necessidade de medicao adicional. As perguntas realizadas para saber se os voluntrios tiveram algum evento adverso foram limitadas a perguntas gerais, tais como: Como vai voc?
Definies de efeitos adversos quanto intensidade
Leve - Experincia adversa facilmente tolerada
Moderada - Experincia adversa desagradvel o bastante para interferir na atividade cotidiana.
Severa - Experincia adversa que impossibilita a realizao da atividade cotidiana normal.
Relacionamento suposto com a droga experimental
Sem relao - A experincia adversa definitivamente no est relacionada droga em estudo.
Desconhecida - H outras causas mais provveis e no h suspeitas de que a droga seja a causa.
104
Possvel - No foi demonstrado um relacionamento de causa e efeito direto entre a droga e a experincia adversa, porm, h uma possibilidade razovel de que a droga esteja envolvida.
Provvel - H um relacionamento direto de causa e efeito entre a experincia e a droga em estudo.
1.12 Aspectos ticos
O projeto de pesquisa, com o protocolo experimental e o termo de consentimento, foram submetidos ao Comit de tica em Pesquisa com Seres Humanos da Universidade Federal do Cear, credenciado pelo CONEP - Conselho Nacional de Sade/MS.
O Estudo foi conduzido de acordo com a Declarao de Helsinque (1964) e as revises de Tquio (1975), Veneza (1983), Hong Kong (1989), Somerset Oeste (1996) e Edimburgo (2000), assim como as regulamentaes locais (Resolues 196/96 e 251/97 do CNS-MS, bem como Resoluo 135/03 da ANVISA).
1.13 Termo de consentimento livre e esclarecido
Segundo a Declarao do Congresso Nacional de Biotica (SIBI), realizado em junho de 2000, o art. 11, dedicado aos temas da pesquisa e experimentao, relata que os sujeitos das experimentaes devero dar seu consentimento livre e esclarecido e plenamente informado.
Os voluntrios selecionados para esse estudo receberam explanao sobre a natureza e os objetivos do estudo. Foi enfatizado que o trabalho teria a finalidade de pesquisa e que o voluntrio no poderia esperar qualquer efeito teraputico. O voluntrio tambm tinha conhecimento da sua liberdade para se retirar a qualquer momento do estudo sem que isto lhe cause qualquer prejuzo no seu atendimento junto Unidade de Farmacologia Clnica ou ao Hospital de Messejana. Foi solicitado a cada voluntrio que, caso concordasse com o protocolo
105
de estudo, assine o Termo de Consentimento Livre e Esclarecido para a participao do estudo.
1.14 Confidencialidade
Os resultados da avaliao mdica, o eletrocardiograma e os exames laboratoriais foram registrados em folha individual de cada voluntrio. Uma cpia dos exames laboratoriais realizados no perodo pr e ps-estudo foi fornecida aos voluntrios, quando solicitada.
2 PROTOCOLO EXPERIMENTAL PARA QUANTIFICAO DOS ANALITOS
2.1 Sistema de Qualidade e Biossegurana
Os principais benefcios de sistemas de gesto da qualidade em laboratrios de pesquisa so: maior credibilidade nos resultados e pesquisadores, comparabilidade e reprodutibilidade de resultados publicados, aumento da confiana entre as partes envolvidas e facilidade na aquisio de financiamentos.
Para garantir que os estudos realizados atendam s expectativas e exigncias das autoridades regulamentadoras e das organizaes que fornecem reconhecimento, a Unidade de Farmacologia Clnica tem seu escopo de trabalho e Sistema de Qualidade baseados nas Normas e Resolues: NBR ISO/IEC 17025:2001; Resoluo ANVISA n 41, de 28 de abril de 2000; Resoluo ANVISA n 133, de 29 de maio de 2003; Resoluo ANVISA n 135, de 29 de maio de 2003; Procedimento GGLAS n 02/17025 e Procedimento GGLAS n 02/BPL; Boas Prticas Clnicas.
O objetivo da biossegurana adotada reconhecer fontes de perigo, avaliar as situaes de risco que essa fonte oferece e control-la, tomando decises tcnicas e/ou administrativas para promover mudanas. Ao adotarmos procedimentos especficos para evitar ou minimizar os riscos de atividades potencialmente perigosas, estamos aplicando a biossegurana.
106
Jaleco, culos de segurana e luvas de procedimentos; so de uso obrigatrio durante os procedimentos e quando for necessria a manipulao de solues biolgicas. Procedimentos de segurana utilizados em laboratrios bem como procedimentos adequados para descarte de lixo qumico e biolgico so seguidos no desenvolvimento dos ensaios.
2.2 Substncias qumicas e Materiais Utilizados
As substncias analticas, cefadroxil e ciprofloxacino foram grau USP, enquanto que a amoxicilina, norfloxacino, carbamazepina, oxcarbazepina e 10,11dihidro-10-hidroxicarbamazepina (MHD) foram obtidos pela Farmacopia Brasileira, Sigma-Aldrich, CDN Isotopes, Ach Laboratrios Farmacuticos S/A e Synefine Researsh, respectivamente, todos com pureza maior que 98% (ANEXOS F, G, H, I, J, L e M). Acetonitrila, diclorometano, metanol, cido actico, cido clordrico, dietil ter e diclorometano foram grau HPLC. Fosfato de potssio monobsico e dibsico, cido ctrico, hidrxido de sdio, cido frmico foram grau analtico. Foi utilizada gua purificada do sistema Milli-Q (Milipore EUA). Amostras de plasma humano foram obtidas do Centro de Hematologia e Hemoterapia do Cear para utilizao em teste de especificidade dos mtodos (Cear, Brasil).
Tabela 1. Utenslios utilizados no desenvolvimento e validao dos mtodos.
Utenslios Micropipetas ajustveis Repipetador Gilson Repipetador Eppendorf Ponteiras para micropipetas ajustveis Tubos plsticos de 50 e 15 mL Tubos de vidro de 120 x 12 mm Misturador Vortex Balana Analtica Mettler Microvial de 300 uL Banho Ultra-snico Produtor, Pas Gilson, Frana Gilson, Frana Eppendorf, EUA Alfa-Lab, Brasil TTP, Sua Laborglass, Brasil Phoenix, Brasil Toledo, Sua OCP Diagnostics, Brasil Odontobrs, Brasil
107
2.3 Padro interno
A escolha dos padres internos utilizado nos processos de validao e desenvolvimento dos mtodos bioanalticos propostos deu-se atravs da anlise de sua estrutura qumica, bem como do frmaco em estudo. Os critrios utilizados para a escolha dos padres internos foram: presena de grupos funcionais similares em ambas s estruturas; semelhana quanto s caractersticas qumicas de cada molcula; e composio qumica elementar. Atravs desta anlise preliminar foi escolhido, como padres internos, o cefadroxil, ciprofloxacino e carbamazepina.
2.4 Preparo das solues padro
As solues mster (1mg/mL) foram preparadas em uma proporo de metanol-gua (1:1) para amoxicilina e cefadroxil; em NaOH 0,01M para norfloxacino e ciprofloxacino; em acetonitrila para oxcarbazepina, MHD e carbamazepina. As solues de trabalho foram obtidas atravs de diluies das solues mster em metanol-gua, gua Milli-Q ou com acetonitrila-gua (50:50 v/v), para os respectivos estudos. Os padres analticos em plasma foram preparados adicionando volumes especficos das solues de trabalho em plasma livre de interferentes obtendo-se concentraes finais na faixa de 0,5-40 g/mL, 30 3500 ng/mL e 20/40 5250/10500 ng/ml, para os respectivos estudos. As solues de controle de qualidade foram fixadas em 1,5, 15 e 30 g/mL, 90, 1400 e 2800 ng/mL e 60/120, 2000/4000 e 4000/8000 para ng/mL (correspondendo da baixa, mdia e alta e concentrao), quantificao amoxicilina, norfloxacino
oxcarbazepina/MHD, respectivamente, e preparadas com as mesmas amostras de plasma utilizadas no preparo da curva de calibrao. As solues de trabalho do padro interno (400 g/ml, 5 g/mL e 400 ng/mL) foram preparadas utilizando soluo de metanol-gua, gua Milli-Q e acetonitrila-gua (50:50 v/v), para cefadroxil, ciprofloxacino e carbamazepina, respectivamente. Todas as solues foram estocadas a 4C e trazidas temperatura ambiente antes do uso.
108
2.5 Critrios para validao do mtodo bioanaltico
2.5.1 Determinao do limite de quantificao (LQ)
O limite de quantificao do mtodo analtico foi definido levando-se em considerao a sensibilidade, especificidade, preciso e exatido do mtodo.
Para a determinao do LQ, foram feitas anlises da matriz biolgica contendo concentraes decrescentes do frmaco at o menor nvel quantificvel com preciso e exatido aceitveis. Com isto, tentou-se assegurar que os menores valores possveis fossem analisados, por meio de repeties de anlises, com valores sucessivamente menores, quando os inicialmente escolhidos fossem quantificados com preciso e exatido aceitveis.
A determinao do LQ do mtodo proposto seguiu os seguintes critrios:
a) A resposta de pico para o LQ deve ser, no mnimo, 5 vezes maior que qualque interferncia na amostra branco no tempo de reteno do frmaco;
b) O pico de resposta do frmaco no LQ deve ser identificvel e reprodutvel com preciso de 20% e exatido entre 80-120% em relao concentrao nominal do padro, atravs da anlise de, no mnimo, cinco amostras dos padres.
2.5.2 Curva de calibrao/linearidade
Para a determinao da curva de calibrao analisaram-se amostras extradas da matriz biolgica em seis concentraes distintas. A curva de calibrao incluiu a anlise da amostra branco (matriz biolgica isenta de padro interno e do padro do frmaco), amostra zero (matriz biolgica mais o padro interno) e mais seis amostras contendo padro do frmaco e padro interno contemplando o limite
109
de variao esperado. As amostras contendo padro do frmaco e padro interno foram analisadas em triplicata.
As concentraes dos padres foram definidas em testes preliminares, incluindo a primeira quantificao de amostras, levando-se em considerao a sensibilidade do mtodo e a faixa prevista das concentraes das amostras a serem determinadas.
A linearidade da curva de calibrao foi avaliada dentro dos seguintes critrios:
a) Desvio menor ou igual a 20% em relao concentrao nominal do LQ, em pelo menos duas das triplicatas;
b) Desvio menor ou igual a 15% em relao a concentrao nominal para as outras concentraes da curva de calibrao, em pelo menos duas das triplicatas;
c) No mnimo 4 de 6 concentraes da curva de calibrao devem cumprir com os critrios descritos, incluindo o LQ e a maior concentrao da curva de calibrao;
d) O coeficiente de correlao ser aceito se for igual ou maior do que 0,98.
2.5.3 Preciso e exatido
Para avaliar a preciso e a exatido do mtodo bioanaltico, quatro concentraes distintas, CQB, CQM, CQA e LQ foram analisadas. Cada amostra foi analisada em quintuplicata. A preciso e a exatido foram determinadas em um mesmo lote (preciso e exatido intralote) e em lotes diferentes (preciso e exatido interlotes). As anlises foram realizadas em trs baterias para cada parmetro.
110
2.5.3.1 Preciso e exatido intralote
A preciso e exatido intralote foram definidas segundo os seguintes critrios:
a) Preciso: para os controles CQB, CQM e CQA o coeficiente de variao (CV) no deveria exceder 15%, e para o LQ admitiram-se valores menores ou iguais a 20%;
b) Exatido: o valor da exatido para as amostras CQB, CQM e CQA no deve exceder 15% do valor nominal e para o LQ admitiu-se desvios menores ou iguais a 20%.
2.5.3.2 Preciso e exatido interlote
A preciso e a exatido interlote seguiu os mesmos critrios de validao para a preciso e exatido intralote.
2.5.4 Especificidade
Para avaliar a especificidade, foram analisadas amostras de plasma humano branco obtidos de seis indivduos do Centro de Hematologia e Hemoterapia do Cear. Cada amostra branco foi testada utilizando os procedimentos de extrao e as condies cromatogrficas propostas, para avaliar interferncia no tempo de reteno do frmaco ou do padro interno e os resultados foram comparados com aqueles obtidos com uma soluo padro dos analitos (amoxicilina, norfloxacino, oxcarbazepina ou MHD) em concentrao prxima ao Limite de quantificao.
2.5.5 Recuperao
O clculo da recuperao do frmaco feito comparando-se as reas dos picos do analito (controles de qualidade baixo, mdio e alto) das amostras extradas
111
do plasma com as reas dos picos do analito preparado em soluo (soluo no extrada). As reas correlacionadas com a concentrao, considerando-se que as reas obtidas para os controles baixo, mdio e alto, preparados em soluo (amostras no extradas) correspondem, respectivamente, as concentraes nominais dos analitos.
O clculo da recuperao do padro interno feito de maneira anloga, ou seja, comparando-se as reas do pico do padro interno extrada do plasma com as reas dos picos do padro interno preparada em soluo (amostra no extrada) na concentrao definida.
2.5.6 Estudo de estabilidade do frmaco no fluido biolgico
Os procedimentos de estabilidade avaliaram a estabilidade do analito no plasma humano sob condies distintas de temperatura e acomodao, bem como sua estabilidade em solues de estoque.
A estabilidade nos fluidos biolgicos dependente das condies de armazenamento, das propriedades qumicas da droga, da matriz empregada (plasma humano) e sistema de armazenamento. O resultado do presente teste relevante somente sob as mesmas condies e no deve ser extrapolado para outras matrizes e sistemas de armazenamento.
Para o teste de estabilidade, uma srie de amostras padro foi preparada da soluo estoque (preparada recentemente no mesmo solvente utilizado para a anlise). Os respectivos controles de qualidade baixo, mdio e alto, incluindo todos os analitos e padres internos, foram usados. Amostras do plasma humano de cada concentrao foram preparadas em volume suficiente para ter alquotas mltiplas. Trs alquotas de cada concentrao foram processadas e quantificadas imediatamente para fornecer valores de referncia (frescos) e outras trs alquotas para cada concentrao foram processadas para os testes desejados.
112
2.5.6.1 Estabilidade no tempo e condies de anlise (curta durao)
Para avaliar a estabilidade do frmaco no tempo e condies de anlise, as amostras processadas foram mantidas dentro do auto-injetor temperatura ambiente. Cada amostra de controle de qualidade foi analisada em triplicata nos tempos determinados a qual excede o tempo em que as amostras poderiam ser mantidas em temperatura ambiente depois de serem descongeladas e antes de serem analisadas. Os resultados foram comparados com aqueles obtidos da anlise de amostras recm-preparadas.
2.5.6.2 Estabilidade do frmaco em ciclos de congelamento e degelo
Congelamento (-20oC) por 24h
Degelo 1
EQUIPAMENTO
Congelamento (-20C) por 24h Degelo 2 Anlise
Extrao
Congelamento (-20C) por 24h
Degelo 3
Figura 12. Esquema dos ensaios de estabilidade em ciclos de congelamento de descongelamento
Para avaliar a estabilidade dos analitos, durante trs ciclos de congelamento e descongelamento, foram analisadas cinco amostras de cada controle de qualidade nas seguintes condies: Degelo 01 - as amostras foram
113
congeladas a 20C e mantidas nesta temperatura por 24 horas. Degelo 02 - Aps este tempo, as amostras foram submetidas ao descongelamento natural, temperatura ambiente, extradas e imediatamente analisadas. Depois de completamente descongeladas, as amostras foram novamente congeladas a 20C, mantidas nesta temperatura por mais 24 horas, descongeladas, extradas e analisadas. Degelo 03 - O mesmo procedimento foi repetido mais uma vez, com o intuito de completar o 3 ciclo de congelamento e descongelamento (Figura 12).
2.5.6.3 Estabilidade de longa durao
Para avaliar a estabilidade do frmaco, em plasma, em condies de longa durao, o tempo de armazenamento deve exceder o intervalo de tempo compreendido entre a data da primeira coleta de amostra e a anlise da ltima amostra dos voluntrios (Figura 13).
EQUIPAMENTO
Anlise Degelo Extrao Congelamento (-20C) por 39, 44 ou 30 dias
Figura 13. Esquema dos ensaios de estabilidade de longa durao.
Para verificao desta estabilidade, utilizaram-se cinco amostras de cada controle de qualidade determinadas durante o processo de validao da metodologia analtica proposta. As concentraes de todas as amostras de estabilidade foram comparadas com as concentraes de amostras recm-preparadas.
114
2.6 Desenvolvimento e validao do mtodo para quantificao da amoxicilina
2.6.1 Equipamentos e condies do HPLC
As
medidas
cromatogrficas
foram
obtidas
usando
equipamento
Shimadzu (Kyoto, Japo), composto por uma bomba LC-10AD, um auto-injetor SIL10AD e um detector diodo SPD-M10A controlado pelo software Class VP6.3 para aquisio dos dados. A separao analtica foi realizada por uma coluna Gemini C18 5 m (150 X 4,6 mm), (Phenomenex, EUA). A fase mvel consistiu de um tampo fosfato de 0,01 M (pH 3,5)/acetonitrila (95:5 v/v) liberada a uma taxa de fluxo de 1,5mL/min em temperatura ambiente. O comprimento de onda utilizado para deteco foi de 203 nm. As amostras foram introduzidas usando o auto-injetor e o volume de injeo foi de 30 l.
2.6.2 Preparao de amostras e processo de extrao
As amostras de plasma foram contaminadas em quantidades definidas de amoxicilina obtendo concentraes finais de 0,5, 1, 2, 4, 20 e 40 g/mL. Um volume de 20 l de soluo de padro interno com concentrao de 400 g/mL e 200 l da soluo de 0,1M de cido ctrico foram adicionados 400 l de amostras de plasma (contaminados com analito) ou de voluntrios, contidos em tubos testes com tampa rosqueada. A soluo resultante foi agitada por 20 segundos, sendo posteriormente adicionadas 800 l de acetonitrila e agitada novamente por 40 segundos. Aps a centrifugao em 5500 rpm por 5 minutos, a soluo do sobrenadante foi transferida para um tubo de vidro e acrescentado 2 mL de diclorometano. As amostras foram agitadas por 40 segundos e centrifugadas (5 minutos, 5500 rpm). A fase aquosa foi transferida para vials e alquotas de 30 l foram injetadas no HPLC. As amostras foram mantidas temperatura ambiente no auto-injetor.
115
2.6.3 Validao do mtodo bioanaltico
As curvas de calibrao foram definidas pela relao entre concentrao da amostra e resposta do equipamento, utilizando-se trs corridas em duplicatas com amostras de plasma contaminadas nas concentraes 0,5, 1, 2, 4, 10, 20 e 40 g/mL. As curvas de calibrao foram construdas pela razo das reas dos picos da amoxicilina pelo padro interno (cefadroxil). Utilizando a regresso linear (fator: 1/x2) foi determinada a inclinao da curva, intercept e coeficiente de correlao.
Os controles de qualidade contendo amoxicilina (1,5, 15 e 30 g/mL) foram utilizados para determinar a preciso intra e interlote no processo de validao e calculados atravs de trs corridas analticas por interpolao das curvas. Oito replicatas de cada controle de qualidade foram determinadas em cada corrida analtica.
A seletividade foi avaliada comparando os cromatogramas de seis amostras de plasma branco com amostras de plasma contaminado com amoxicilina e padro interno. A determinao do menor limite de quantificao, definida como a menor concentrao analisada com exatido e preciso, de grande importncia em estudos farmacocinticos. O limite de quantificao de concentrao de 0,5 g/mL foi calculado utilizando oito replicatas. A especificidade do estudo foi determinada utilizando diferentes amostras de plasma. A recuperao absoluta da amoxicilina em plasma foi determinada comparando as reas dos picos obtidos nos trs nveis de amostras de plasma contaminada (1,5, 15 e 30 g/mL) com as obtidas depois da injeo direta de amoxicilina dissolvida em fase mvel nas mesmas concentraes, atravs do sistema cromatogrfico. A estabilidade da amoxicilina foi investigada sob uma variedade de condies de processos e estocagem. Os estudos de adequao do sistema tambm foram realizados e os parmetros como eficincia da coluna, resoluo e assimetria de picos foram avaliados.
116
2.7
Desenvolvimento
validao
do
mtodo
para
quantificao
do
norfloxacino
2.7.1 Equipamentos e condies do HPLC
As medidas cromatogrficas foram obtidas usando um equipamento Shimadzu (Kyoto, Japan), composto por uma bomba LC-10AD, um autoinjetor SIL10AD e um detector fluorimtrico RF-10Axl configurado com 300 e 450 nm de excitao e emisso, respectivamente. O sistema foi operado utilizando o programa Class VP 6.3 para aquisio dos dados. A separao foi realizada em coluna analtica Synergi MAX-RP 4m (150x4,6 mm) (Phenomenex, Torrance, CA, USA) sem pr-coluna. A fase mvel consistiu de uma soluo de acetonitrila-fosfato de potssio monobsico (pH, 3.0; 25 mM; 15:85 v/v), eludo em um fluxo de 1,0 mL/min. O pH do tampo fosfato foi ajustado com cido fosfrico. A coluna foi operada em 40C.
2.7.2 Preparao de amostras e processo de extrao
Para elaborao dos padres de calibrao e amostras desconhecidas, o plasma (200 L) foi armazenado em tubo apropriado contendo 50 L de padro interno de 5 g/mL e 1,6 mL de acetonitrila. Os tubos foram agitados vigorosamente por 40 segundos para ocorrer desproteinizao e ento centrifugados em 3500g por 10 minutos. O sobrenadante foi separado e evaporado (50C) sob fluxo de nitrognio. O resduo foi reconstitudo em 200 L de tampo fosfato (25 mM), as amostras foram transferidas para tubos vials e inseridas no autoinjetor 8C, sendo injetada no sistema cromatogrfico.
2.7.3 Validao do mtodo bioanaltico
As curvas de calibrao so definidas pela relao entre concentrao da amostra e resposta do equipamento, utilizando-se trs corridas em duplicatas com amostras de plasma contaminadas, nas concentraes (30, 500, 1000, 1500, 2000,
117
2500, 3000 e 3500 ng/mL). As curvas de calibrao foram construdas pela razo das reas dos picos do norfloxacino pelo padro interno. Atravs da regresso linear (fator: 1/x) foi determinada a inclinao da curva, intercept e coeficiente de correlao. Os controles de qualidade contendo norfloxacino (90, 1400 e 2800 ng/mL) foram utilizados para determinar a preciso intra e interlote no processo de validao e calculados atravs de trs corridas analticas por interpolao das curvas. Oito replicatas de cada controle de qualidade foram determinadas em cada corrida analtica.
O limite de quantificao (LQ) de concentrao foi calculado utilizando oito replicatas. A especificidade do estudo foi determinada utilizando seis diferentes amostras de plasma. Cada amostra foi testada para verificar a presena de interferentes com a utilizao do procedimento proposto de extrao e condies cromatogrficas, comparando com aqueles obtidos em soluo aquosa contendo analito em concentrao prxima ao LQ.
A eficincia no processo de extrao do norfloxacino e ciprofloxacino em 3 lotes diferentes de plasma foi determinada por anlise das amostras de controle de qualidade. As amostras foram extradas de acordo com o processo descrito anteriormente. A recuperao relativa foi determinada comparando as mdias das reas de 5 amostras de plasma de cada controle de qualidade contaminadas antes e aps a extrao (n = 5). De acordo com Matuszewski e colaboradores, este procedimento leva em considerao o efeito matriz, obtendo uma recuperao real. A recuperao do padro interno foi tambm determinada pelo mesmo procedimento.
A estabilidade do norfloxacino em trs concentraes (90, 1400 e 2800 ng/mL) foi investigada sob diversas condies de tempo e temperatura. A estabilidade do norfloxacino e ciprofloxacino foi determinada depois de 92 horas em amostras extradas e estocadas no autoinjetor (8C). A estabilidade por congelamento e descongelamento de amostras de plasma foi conduzida em 3 ciclos de congelamento e degelo durante 3 dias. A estabilidade de curta-durao depois de 15 horas de armazenamento em temperatura ambiente (20C) tambm foi avaliada
118
em amostras de controle de qualidade. Alm disso, estabilidade longa-durao depois de 44 dias de armazenamento (em -20C) foi determinada. Todas as amostras (n=5) foram analisadas utilizando amostras para curva de calibrao recm preparadas.
2.8
Desenvolvimento
validao
do
mtodo
para
quantificao
da
oxcarbazepina
2.8.1 Equipamentos e condies do HPLC
As medidas cromatogrficas foram obtidas usando um equipamento Shimadzu (Kyoto, Japan), composto por uma bomba LC-10AD, um autoinjetor SIL10AD. A fase mvel consistiu de acetonitrila-gua (50:50 v/v) + 20 mM cido actico em um fluxo de 1 mL/min atravs da coluna analtica Phenomenex Luna C18 5 m (150x4,6 mm), em temperatura ambiente. Um split da coluna eluente de aproximadamente 1:10 foi inserido, entrando no espectrmetro de massa somente 50 l/minuto. O volume de injeo foi de 5l. O espectrmetro de massa (Micromass Quattro LC) foi equipado com uma fonte de ionizao eletrospray no modo positivo (ES+) e configurado em monitoramento de mltiplas reaes (MRM), monitorando as transies m/z 253 > 208, m/z 255 > 194 e m/z 247 > 204 para oxcarbazepina, MHD e carbamazepina (padro interno), respectivamente. A fonte e a temperatura de dessolvatao foram de 120 e 350 C, respectivamente. O cone e fluxo do gs de dessolvatao (N2) foi de 107 e 353 l/h, respectivamente. O dwell time foi configurado em 0,5 segundos, os valores de voltagem capilar, a energia do cone e energia de coliso foram de 3 kV, 15V, 15 eV para oxcarbazepina, 3 kV, 20V, 20 eV para MHD e 3 kV, 23V, 15 eV para carbamazepina, com presso de gs (argnio) de 2.83103 mbar. A aquisio e anlise dos dados foram obtidas usando o programa MassLynx (V 4.0 em Windows XP, Pentium PC).
2.8.2 Preparao de amostras e processo de extrao
Uma alquota de 25 l de padro interno (400 ng/ml) e 100 l de gua foram adicionadas em 100 l de plasma e agitados. Em seguida, foi adicionado 1 ml
119
de dietil ter-diclorometano (60:40 v/v) nos tubos e agitados em vortex por 5 minutos. Ento, os tubos foram centrifugados em 2000g por 5 minutos em 8C. A parte superior orgnica foi cuidadosamente removida, transferidos para novos tubos e evaporados sob fluxo de nitrognio 50C. O resduo foi reconstitudo em 100 l de acetonitrila seguido de agitao por 30 segundos, transferido para vials e injetado no sistema cromatogrfico.
2.8.3 Validao do mtodo bioanaltico
O procedimento descrito foi validado de acordo com recomendaes internacionais para mtodos bioanalticos (HUBERT; CHIAP, 1999; SHAH et al., 2000). As curvas de calibrao so definidas utilizando-se quatro corridas de diferentes lotes de plasma em triplicatas com amostras de plasma contaminadas nas concentraes (20, 750, 1500, 2250, 3000, 3750, 4500 e 5250 ng/ml para oxcarbazepina e 40, 1500, 3000, 4500, 6000, 7500, 9000 e 10500 ng/ml para MHD). As curvas de calibrao foram construdas pela razo das reas dos picos dos analitos pelo padro interno. O menor limite de quantificao (LQ) foi definido como a menor concentrao a qual pode ser quantificada com um valor de rudo abaixo de 20% e relao sinal-rudo menor que 15 (n=5). O efeito matriz foi determinado em seis diferentes lotes de plasma branco (VAN ROOYEN et al., 2002). Cada amostra branco foi testada para verificar interferncias utilizando o procedimento de extrao e comparados com aqueles obtidos com soluo aquosa do analito em uma concentrao prxima do LQ. A seletividade do sistema de deteco foi verificada para avaliar a possibilidade de cross-talk. Este teste foi realizado com as seguintes etapas: (1) injeo de branco extrado e monitorando a resposta na transio oxcarbazepina, MHD e carbamazepina; (2) injetando separadamente uma amostra de plasma contaminada somente com oxcarbazepina ou MHD na concentrao mais alta e monitorando a resposta na transio do padro interno e MHD ou oxcarbazepina; (3) injetando uma amostra de plasma contaminada somente com padro interno e monitorando a transio da oxcarbazepina e MHD. A preciso e exatido inter e intralote do mtodo foram avaliadas realizando analises em replicatas (n=8) das amostras em plasma dos controles de qualidade com curva de calibrao. O procedimento foi repetido em diferentes dias (n=4) para determinar a
120
preciso e exatido interlote dos dados. A exatido e preciso intralote foram determinadas atravs de amostras contaminadas em replicatas no mesmo dia. A exatido (percentual do erro relativo (%ER)) foi calculada pela razo da concentrao experimental e a nominal das amostras de plasma contaminadas. A preciso do mtodo (anlise inter e intralote) foi calculada pelo percentual do desvio padro relativo (DPR%) entre as medidas repetidas.
A eficincia do mtodo de extrao para oxcarbazepina e MHD em trs lotes diferentes de plasma foi determinada analisando as amostras do controle de qualidade. A recuperao relativa foi determinada comparando a mdia das reas ou respostas de cinco amostras de plasma de cada controle contaminadas antes da extrao com as contaminadas aps a extrao (n=5). A recuperao do padro interno foi tambm determinada pelo mesmo procedimento.
A estabilidade da oxcarbazepina e MHD estocado em trs concentraes foi investigada sob diversas condies de tempo e temperatura. A estabilidade em amostras extradas e estocadas no autoinjetor (8C) foi determinada depois de 92 horas. A estabilidade por congelamento e descongelamento de amostras de plasma foi conduzida com 3 ciclos de congelamento e degelo durante 3 dias. A estabilidade curta-durao para amostras de controle de qualidade depois de 15 horas de armazenamento em temperatura ambiente (20C) tambm foi avaliada. Alm disso, a estabilidade longa-durao depois de 30 dias de armazenamento (em -20C) foi determinada. Todas as amostras (n=5) foram analisadas utilizando amostras para curva de calibrao recm preparadas.
2.9 Aplicao dos mtodos propostos em estudos farmacocinticos
Para avaliao dos parmetros farmacocinticos tais como rea sob a curva do tempo zero ao infinito (AUC0-), do tempo zero a ltima coleta (AUC0-h), concentrao mxima atingida (Cmax), tempo para atingir a concentrao mxima (Tmax), meia-vida plasmtica (t1/2) foram calculados.
121
A meia-vida foi calculada atravs da frmula (t1/2 = ln2/K). A concentrao plasmtica mxima observada (Cmax) e o tempo para obter esta concentrao (Tmax) foram obtidos diretamente da curva. A rea sob a curva de concentrao plasmtica versus tempo de (AUC0-h) foi calculada atravs do mtodo trapezoidal. A extrapolao dessa rea para o infinito (AUC[0-]) foi feita adicionando o valor da relao C0-h/K (onde C0-h a concentrao plasmtica obtida da ltima coleta e K, que a constante de eliminao) a AUC0-h.
2.10 Anlise Estatstica
As
anlises
estatsticas
foram
conduzidas
aps
transformao
logartmica, baseada em modelo aditivo para todos os valores de AUC e Cmax. Tmax, t, K e C0-h no sero avaliados estatisticamente.
Foi empregada a anlise de varincia apropriada para o modelo de 2 perodos cruzados, sob os dados de AUC e Cmax transformados logaritmicamente, a qual levar em conta em seu modelo os efeitos de seqncia, voluntrio dentro da seqncia, tratamento e perodo. Para o delineamento crossover 2x2, o objetivo da anlise de varincia estudar a variabilidade de seqncia e perodo dos dados estudados.
A verificao de existncia de efeito residual foi realizada com base nos testes de seqncia da ANOVA, utilizando-se como parmetro o P_value, obtido como base na F_stat do efeito de seqncia (Sequence Hypothesis of Model Efects).
Foram calculados os pontos paramtricos e estimativa dos intervalos da razo T/R (formulao "teste" / formulao "referncia") para valores AUC e Cmax. A biodisponibilidade relativa da formulao Teste versus Referncia foi avaliada pelas razes das mdias geomtricas (pontos estimados). O intervalo de confiana de 90% serviu como estimativa de intervalo e foi determinado por anlises paramtricas (dois testes t unicaudais p=0.05).
122
Os medicamentos so considerados bioequivalentes quando IC de 90% para a razo entre as mdias dos dados transformados em logaritmo natural de Cmax e ASC0-ulth estiverem entre 80 e 125%, conforme legislao vigente (FDA, 1985; FDA, 1993).
A anlise realizada com o emprego dos seguintes softwares: WinNonLin Professional Network Edition, Verso 5.0; Bioequivalence Program for Two-Period Crossover Studies - Verso 3.4, por Jerman P Wijnand; Microsoft Office Excel 2003 (11.5612.5606), MedCalc Verso 7.2.1.0 e Graph Pad Prism Verso 3.02.
123
PARTE III: RESULTADOS E DISCUSSO
124
PARTE III: RESULTADOS E DISCUSSO
CAPTULO 1: Amoxicilina
Amoxicilina (4-amino-5-cloro-N-[(2-dietilamino) etil]-2-metoxi benzamida) um dos mais comuns antimicrobianos, possui largo espectro de ao e pertence ao grupo das penicilinas. freqentemente prescrito na prtica mdica e veterinria, individualmente ou combinado com outros frmacos, para o tratamento sistmico de infeces (YUAN et al., 1995; DOUSA; HOSMANOVA, 2005). A co-administrao de omeprazol (inibidor de bomba de prtons) com amoxicilina tem sido amplamente usada para controlar os sintomas de gastrite e lcera pptica associada infeco de Helicobacter pylori (ERAH et al., 1997).
Vrios mtodos, incluindo microbiolgicos (BALL et al., 1980) e ensaio enzimtico (CULLMANN; DICK, 1986), foram utilizados para quantificar a concentrao de amoxicilina nos fluidos biolgicos, individualmente e em combinao com outros frmacos. Alguns deles, como bioensaio, requerem tempo e no so especficos (MENELAOU et al., 1999). Assim, quando critrios tais como a sensibilidade e especificidade precisam ser asseguradas, geralmente, utilizada a cromatografia lquida de alta eficincia (HPLC). Ao longo dos anos, vrios mtodos cromatogrficos com deteco fluoromtrica ou ultravioleta tm sido desenvolvidos para anlise de amoxicilina (YUAN et al., 1995; DOUSA; HOSMANOVA, 2005; MENELAOU et al., 1999; HOIZEY et al., 2002; WIBAWA et al., 2002). Mais recentemente, a separao por HPLC com deteco por espectrometria de massa tornou-se um mtodo de escolha (KYUANG-HWAN, 2004). Com exceo ao HPLC/MS, estes mtodos geralmente requerem uma extrao em fase slida, como processo de preparao das amostras, a fim de aumentar a sensibilidade e seletividade. Embora esses mtodos sejam aplicados com sucesso, nem sempre so simples e, em muitos casos, ambos so morosos e dispendiosos.
Para compreender o comportamento farmacocintico da Amoxicilina em seres humanos, foi necessrio utilizar um mtodo quantitativo confivel. Foram
125
desenvolvidos diversos mtodos de cromatografia para deteco de amoxicilina em fludos biolgicos, a maioria com deteco em ultravioleta (UV) utilizando comprimentos de onda baixos (= 210 a 330 nanmetro). Aps a avaliao de vrias condies cromatogrficas, um mtodo apropriado e simples para quantificao de amoxicilina em plasma humano foi desenvolvido utilizando cromatografia lquida de alta eficincia com deteco por ultravioleta (UV).
1 ETAPA CLNICA
O estudo foi delineado, de forma a permitir que se obtenham os parmetros farmacocinticos relevantes para a comparao estatstica, visando a averiguao de biodisponibilidade. No caso em estudo, tais parmetros foram obtidos diretamente a partir da determinao da concentrao plasmtica de norfloxacino, baseado na aplicao de um modelo no-compartimental prprio para avaliao destas concentraes, aps a administrao do medicamento por via oral.
Por conseguinte, a finalidade primria da Etapa Clnica foi a coleta de amostras de sangue dos voluntrios para medir (na Etapa Analtica) nveis plasmticos das substncias aps sua administrao oral.
O desenho consistiu de um estudo aberto, randomizado, com replicao, de 24 voluntrios sendo doze homens e doze mulheres no grvidas. O nmero de voluntrios sadios deve sempre assegurar poder estatstico suficiente para garantir a confiabilidade dos resultados do estudo (BRASIL, 2003b). O termo voluntrio normal ou sadio deve ser interpretado como indivduo adulto, sem anormalidades clnicolaboratoriais capazes de prejudicar a interpretao do experimento ou aumentar a sensibilidade do indivduo ao potencial txico do frmaco em estudo. A idade variou entre 20 e 47 anos (27,33 9,15 anos), altura variando de 151 a 176 cm (164,30 8,21 cm) e ndice de massa corporal mdio de 24,12 2,65 kg/m2.
Os voluntrios qualificados para participar do estudo foram internados por 02 perodos de aproximadamente 24 horas, com 3 dias de intervalo entre os internamentos, para garantir que no houvesse um efeito residual do primeiro
126
frmaco administrado, que poderia inflar ou alterar artificialmente os resultados do segundo e assim por diante (HOWARD et al., 2000).
O rgo responsvel pelos medicamentos genricos no Brasil, Agncia Nacional de Vigilncia Sanitria ANVISA, determina que o cronograma de coletas das amostras sanguneas dever contemplar tempo igual ou superior a 3 a 5 vezes meias-vidas de eliminao do frmaco ou metablito ativo (BRASIL, 2003b). Devese incluir o maior nmero possvel de amostras na regio prximo a concentrao mxima atingida para que possibilite a determinao da rea sob a curva de concentrao sangnea (BRASIL, 2003a). Amostras sangneas para a determinao dos nveis plasmticos dos frmacos foram obtidas antes da administrao (tempo zero e pool) e aps a administrao, seguindo o seguinte esquema:
Amoxicilina 20, 40, 60, 90, 120, 150, 180, 240, 360; 480 e 600 minutos.
2 DESENVOLVIMENTO E VALIDAO DO MTODO BIOANALTICO PARA QUANTIFICAO DA AMOXICILINA
2.1 Seletividade do mtodo
Foi desenvolvido um mtodo utilizando cromatografia lquida de alta eficincia com deteco ultravioleta para determinao de amoxicilina em plasma humano. conhecido que a composio da fase mvel, bem como, a preparao das amostras so fatores crticos para produzir uma satisfatria separao dos compostos desejados e impurezas de fluidos biolgicos. Diferentemente de alguns mtodos que requerem tcnicas adicionais do sistema HPLC para quantificao da amoxicilina em plasma durante as primeiras fases de desenvolvimento do mtodo, foram feitas tentativas (composio da fase mvel, preparao das amostras) para produzir boa separao da amoxicilina e padro interno dos compostos endgenos sem haver mudana na tcnica de separao.
127
Os cromatogramas livres de analitos apresentaram ausncia de interferentes endgenos da matriz ao longo da janela de eluio para amoxicilina e padro interno, os picos esto apresentados na figura 14. O tempo de reteno para amoxicilina e padro interno foram 4,2 e 5,5 minutos, respectivamente, que pode ser considerado um tempo curto para corrida analtica. Assim, o tempo de reteno para amoxicilina foi aproximadamente duas vezes mais curto que do mtodo anteriormente publicado (YUAN et al., 1997; MENELAOU et al., 1999; HOIZEY et al., 2002).
128
D et ect o r A - 1 ( 202n m ) B ran co Plasm a N o rm al L o t 0000487682- 2 M ET 006V01T B M 02- 11
1.2
R e ten tio n Time A re a N a me
1. 2
1.0
1. 0
0.8
0. 8
Volts
0.6
0. 6
0.4
0. 4
(amoxicilina)
(cefadroxila)
0.2
0. 2
0.0
0. 0 0 1 2 3 4 5 Min ut e s 6 7 8 9 10 11
Detector A - 1 (202nm) CQA Amoxicilina 30 ug/mL MET006V01VAL03-52
1.2
1.2
Retention Time Area Name
1.0
1.0
0.8
4.175 1567338 amoxicilina
0.8
5.326 1097133 cefadroxila
Volts
0.6
0.4
0.4
0.2
0.2
0.0
0.0 0 1 2 3 4 5 Minutes 6 7 8 9 10 11
Detector A - 1 (202nm) VOL 10 DC280584 - 1 FASE - 01:00 120-04DCV04-026
0.6
Retention Time Area Name
0.6
C
0.5
0.4
0.5
4.109 144798 amoxicilina
5.243 879211 cefadroxila
0.4
Volts
0.3
0.2
0.2
0.1
0.1
0.0
0.0
5 Minutes
10
11
Figura
14.
Cromatograma
representativo
do
plasma
branco
(A),
Volts
0.3
Volts
0.6
Volts
plasma
contaminado com amoxicilina (30 g/mL) e cefadroxil (padro interno) (B) e plasma de um voluntrio aps 1h da administrao oral de amoxicilina contaminado com cefadroxil (C).
129
2.2 Linearidade do mtodo
As curvas de calibrao, definidas em trs corridas analticas em duplicata, foram lineares para a faixa de concentrao de 0,5 - 40 g/mL para amoxicilina em plasma, com um coeficiente de correlao maior que 0,99. A linearidade foi definida pela seguinte equao: y = 19,0642x + 0,000162149, onde y representa a relao da altura do pico da amoxicilina e padro interno e x representa a relao da concentrao (Figura 15).
Figura 15. Representao grfica da curva de calibrao y = 19,0642x + 0,000162149, r2= 0,999253.
O limite de quantificao da amoxicilina foi de 0,5 g/mL, sendo este a menor concentrao a qual pode ser quantificada com desvio abaixo de 20% e relao sinal rudo menor que 5 (n=8). Este limite realmente suficiente para realizar estudos de biodisponibilidade e foi selecionado no que diz respeito concentrao esperada das amostras no estudo de biodisponibilidade.
2.3 Preciso e exatido do mtodo
No estudo, a preciso foi avaliada intra e interlote. O percentual do coeficiente de variao (%CV) foi utilizado como medida de preciso. Os dados para
130
preciso intra e interlote para amoxicilina so apresentados na tabela 2. A validao estatstica apresentou uma boa preciso (abaixo de 10%) para todas as concentraes (1,5, 15 e 30 g/mL) de amoxicilina. Para serem aceitveis, os valores devem estar dentro de 15% em todas as concentraes. Tabela 2. Validao da quantificao de amoxicilina em plasma: preciso intra e interlote das amostras de controle de qualidade (n = 8).
Intralote (n=8) Concentrao Nominal (gml ) Mdia DP Exatido (%) Preciso (%) Interlote (n=8) Mdia DP Exatido (%) Preciso (%) 0,51 0,05 98,0 9,8 1,57 0,08 95,3 5,1 14,50 0,58 96,7 4,0 28,52 1,49 95,1 5,2
-1
LQ 0,5 0,52 0,05 96,0 9,6
CQB 1,5 1,51 0,09 99,3 6,0
CQM 15 14,88 0,39 99,2 2,6
CQA 30 30,32 0,90 98,9 2,9
2.4 Recuperao do mtodo
Os trs nveis de concentrao da amoxicilina (n=5) foram usados para determinar a recuperao absoluta. Foram encontrados bons resultados para os valores mdios da recuperao de amoxicilina em 1,5 g/mL (59,4 3,66%), 15 g/mL (60,5 1,74%) e 30 g/mL (67,1 3,82%), sendo a recuperao global de 62,3%. Valores de recuperao no menores que 50, 80 e 90% tm sido utilizados como limites de aceitao. Embora seja desejvel atingir a recuperao o mais prximo possvel de 100% (KARNES et al., 1991), pode ser aceitvel uma baixa recuperao se for demonstrado um bom resultado reprodutvel nessa faixa. Os resultados do estudo foram reprodutveis e precisos, sendo o desvio padro relativo inferior a 7%.
2.5 Estudos de Estabilidade do mtodo
A determinao da estabilidade da amoxicilina foi realizada analisando amostras de plasma imediatamente, horas ou dias subseqentes do perodo de estoque. Os valores mdios e os desvios (%) calculados de trs nveis de amostras
131
de plasma esto apresentados na tabela 3. Todas as amostras (n=5) foram analisadas usando amostras recm-preparadas da curva de calibrao. As anlises de trs conjuntos de amostras de plasma contaminadas com concentraes baixa (1,5 g/mL), mdia (15 g/mL) e alta (30 g/mL) de amoxicilina estocadas -18C e expostas trs ciclos de congelamento e descongelamento no apresentaram perda significante do analito. Tambm foi analisada a estabilidade das amostras de plasma processado e estocados no autoinjetor temperatura de 23C em um perodo de 59 horas. Os resultados mostraram que as reas dos picos de amoxicilina permaneceram praticamente sem alteraes e picos adicionais no foram observados. Posteriormente, para demonstrar a estabilidade de longa durao da amoxicilina, trs amostras de plasma contaminadas foram processadas e estocadas -20C por 39 dias, sendo encontrada estabilidade neste perodo. Tabela 3 Resultado da estabilidade (mdia desvio padro) obtido com trs amostras de plasma com concentrao baixa (1,5 g/mL), mdia (15 g/mL) e alta (30 g/mL) de amoxicilina.
1,5 g/mL Antes Congelamento e degelo (X DP) Variao (%) Antes Ps-Processamento (X DP) Variao (%) Antes Curta durao (X DP) Variao (%) Antes Longa durao (X DP) Variao (%)
1,47 0,12 1,58 0,14 1,53 0,11 1,42 0,02
15 g/mL Antes
15,30 0,26
30 g/mL Antes
30,93 0,53
3 Ciclo
1,44 0,07
3 Ciclo
15,63 0,14
3 Ciclo
29,81 0,75
1,4 59 h
1,32 0,05
2,2 Antes
15,64 0,39
-3,6 59 h Antes
30,93 0,49
59 h
26,66 0,66
13,82 0,53
-13,7 18 h
1,42 0,03
-11,6 Antes
15,66 1,49
-13,8 18 h Antes
32,83 0,49
18 h
28,92 1,11
14,47 0,31
-10,1 39 dias
1,39 0,08
-7,6 Antes
15,91 0,39
-11,9 39 dias Antes
30,23 0,74
39 dias
29,94 0,33
15,98 0,10
-5,4
0,4
-1,0
132
APLICAO
DO
MTODO
BIOANALTICO
EM
ESTUDO
DE
BIOEQUIVALNCIA ENTRE DUAS FORMULAES CONTENDO AMOXICILINA (CPSULA DE 500MG) EM VOLUNTRIOS SADIOS
O ensaio foi aplicado para avaliar a biodisponibilidade de dois produtos contendo 500 mg de amoxicilina, a fim de determinar se a formulao teste produzida pela Glenmark Pharmaceuticals Ltda e referncia produzida pela GlaxoSmithKline Beecham so bioequivalentes. As curvas de concentrao plasmtica mdia da amoxicilina aps administrao oral da formulao teste e referncia versus tempo esto apresentadas na figura 16. Os parmetros farmacocinticos correspondentes esto descritos na tabela 4. Os resultados foram semelhantes aos j publicados, em que a amoxicilina foi quantificada por RP-HPLC com deteco em espectrmetro de massa (YOON et al., 2004; NASIR et al., 2004) ou deteco UV (BAGLIE et al., 2005). Tabela 4 Parmetros farmacocinticos da formulao teste e referncia (amoxicilina 500 mg) depois da administrao oral de 24 voluntrios.
Formulao Teste
Cmax
(g*mL ) Mdia Mnimo Mximo D.P.
-1
Formulao Referncia
t1/2
-1
Tmax
(h)
AUC0-10h
(g*h*mL )
-1
AUC0-
(g*h*mL )
Cmax (g*mL )
-1
(h)
Tmax (h)
AUC0-10h
(g*h*mL )
-1
AUC0-
(g*h*mL )
-1
t1/2 (h)
5,60 1,45 9,63 1,85 32,97
2,04 1,00 3,00 0,74
36,05
15,30 4,71 22,93 3,94 25,77
15,66 5,03 23,42 3,92 25,04
1,02 0,61 1,90 0,28 26,90
6,10 4,04 9,39 1,25 20,50
1,91 0,67 3,00 0,74 38,74
15,66 9,93 21,82 3,55 22,68
16,25 10,69 22,92 3,58 22,06
0,99 0,66 1,90 0,28 28,48
CV(%)
133
Amoxicilina (ug/mL)
4 3 2 1 0 0 2 4 6
Referncia Teste
10
Tempo (h)
Figura 16. Grfico da curva de concentrao plasmtica (g/dL) versus tempo (h) da amoxicilina. Representa a mdia das concentraes plasmticas obtidas aps a administrao de duas formulaes de Amoxicilina 500 mg em volutrios sadios.
A concentrao plasmtica mxima observada (Cmax) e o tempo para obter esta concentrao (Tmax) foram obtidos diretamente da curva. A rea sob a curva de concentrao plasmtica versus tempo de (AUC0-h) foi calculada atravs do mtodo trapezoidal. A extrapolao dessa rea para o infinito (AUC[0-]) foi feita adicionando o valor da relao C0-h/K (onde C0-h a concentrao plasmtica obtida da ltima coleta e K, que a constante de eliminao) a AUC0-h (Tabela 5).
Tabela 5 - Mdia, IC (90%) e concluso para a razo das mdias de Cmax, AUC0-ult h e AUC0- Dados transformados em logaritmo natural. Razo: Amoxicilina / Amoxil Cmax AUC0-ult h AUC0-
Mdia Geomtrica 87,362 97,388 95,223
IC (90%) (76,46 99,82) (88,00 107,78) (87,05 104,17)
CV (%) 27,362 20,661 18,260
Bioequivalncia No Sim Sim
Na Tabela 5 notamos que o coeficiente de variao intraindividual para Cmax foi de 27,362%, acima de 20%, o que determinou um intervalo de confiana (90%) alm dos limites permitidos (80 a 125%), estando em desacordo com o que preconiza a legislao vigente. A anlise estatstica da AUC0t, AUC0 e Cmax
134
revelaram que no houve bioequivalncia entre as 2 formulaes. A Food and Drug Administration (FDA) propem que formulaes farmacuticas sejam consideradas bioequivalentes quando a razo das mdias de Cmax e AUC(0-t) apresentarem-se entre 80125% (considerando um intervalo de confiana de 90%) (FDA, 1985; FDA, 1993).
CAPTULO 2: Norfloxacino
Norfloxacino
[1-etil-6-fluoro-1,
4-dihidro-4-oxo-7-(1-piperazinil)-3-cido
quinolinocarboxilico] um antimicrobiano fluoroquinolona droga da classe cujo espectro de atividade inclui tanto gram-negativas e bactrias gram-positivas. frequentemente prescrito na prtica mdica para o tratamento de infeces sistmicas. Norfloxacino administrado por via oral, como comprimidos revestidos por pelcula contendo 400 mg. Nesta dose, a concentrao plasmtica mxima (Cmax), varia de 1,6 a 2,87 mg/mL (SETH et al., 1995).
Mtodos cromatogrficos tm sido descritos para a determinao quantitativa do norfloxacino em fluidos biolgicos. A maioria dos mtodos para a determinao quantitativa do norfloxacino inclui a cromatografia lquida de alta eficincia com deteco ultravioleta (CRDOBA-BORREGO et al., 1999; SAMANIDOU et al., 2003; SHERVINGTON et al., 2005; MYERS; BLUMER, 1987; PISTOS et al., 2005) ou de fluorescncia (HUSSAIN et al., 1995; HAN et al., 2005; MASCHER; KIKUTA, 1998; WELL et al., 1998). No entanto, quando os critrios, tais como a sensibilidade e especificidade, necessitam ser assegurados para anlise de rotina, a cromatografia lquida de alta eficincia de fase reversa (RP-HPLC) com deteco fluorimtrica especialmente vantajosa. Esses mtodos, as concentraes de norfloxacino nem sempre eram detectveis em pequenas amostras de plasma (0,5-1 ml). Este fato os tornou imprprios devido o tempo de curso da anlise, nos casos em que pequenos volumes de plasma so utilizados (estudos farmacocinticos experimentais em animais ou crianas). Um menor volume plasmtico foi utilizado por Saramanidou et al. (2003) (50 L) e Hussain et al. (1995) (150 L), mas com tempo de reteno do norfloxacino de 7,4 e 6,9 minutos,
135
respectivamente. Estes tempo de reteno relativamente longos do norfloxacino no so adequados para a quantificao de um nmero elevado de amostras.
O presente mtodo no totalmente diferente daqueles descritos no pargrafo anterior, porm combina menor volume plasmtico, curto tempo de reteno, um simples processo de extrao atravs de precipitao das protenas plasmticas, boa sensibilidade, exatido, preciso e linearidade.
1 ETAPA CLNICA
O estudo foi delineado, de forma a permitir que se obtenham os parmetros farmacocinticos relevantes para a comparao estatstica, visando a averiguao de biodisponibilidade. No caso em estudo, tais parmetros foram obtidos diretamente a partir da determinao da concentrao plasmtica de norfloxacino, baseado na aplicao de um modelo no-compartimental prprio para avaliao destas concentraes, aps a administrao do medicamento por via oral.
Por conseguinte, a finalidade primria da Etapa Clnica foi a coleta de amostras de sangue dos voluntrios para medir (na Etapa Analtica) nveis plasmticos das substncias aps sua administrao oral.
O desenho consistiu de um estudo aberto, randomizado, com replicao, de 24 voluntrios sendo doze homens e doze mulheres no grvidas. O nmero de voluntrios sadios deve sempre assegurar poder estatstico suficiente para garantir a confiabilidade dos resultados do estudo (BRASIL, 2003b). O termo voluntrio normal ou sadio deve ser interpretado como indivduo adulto, sem anormalidades clnicolaboratoriais capazes de prejudicar a interpretao do experimento ou aumentar a sensibilidade do indivduo ao potencial txico do frmaco em estudo. A idade variou entre 18 e 48 anos (28,42 8,29 anos), altura variando de 152 a 177 cm (165,50 7,98 cm) e ndice de massa corporal mdio de 23,44 2,43 kg/m2.
Os voluntrios qualificados para participar do estudo foram internados por 02 perodos de aproximadamente 36 horas, com 7 dias de intervalo entre os
136
internamentos, para garantir que no houvesse um efeito residual do primeiro frmaco administrado, que poderia inflar ou alterar artificialmente os resultados do segundo e assim por diante (HOWARD et al., 2000).
O rgo responsvel pelos medicamentos genricos no Brasil, Agncia Nacional de Vigilncia Sanitria ANVISA, determina que o cronograma de coletas das amostras sanguneas dever contemplar tempo igual ou superior a 3 a 5 vezes meias-vidas de eliminao do frmaco ou metablito ativo (BRASIL, 2003b). Devese incluir o maior nmero possvel de amostras na regio prximo a concentrao mxima atingida para que possibilite a determinao da rea sob a curva de concentrao sangnea (BRASIL, 2003a). Amostras sangneas para a determinao dos nveis plasmticos dos frmacos foram obtidas antes da administrao (tempo zero e pool) e aps a administrao, seguindo o seguinte esquema:
Norfloxacino 20, 40, 60, 80, 100, 120, 150, 180, 240, 360, 480, 600, 720, 1080, 1440 minutos.
2 DESENVOLVIMENTO E VALIDAO DO MTODO BIOANALTICO PARA QUANTIFICAO DA NORFLOXACINO
2.1 Otimizao dos procedimentos gerais
Entre as diferentes tcnicas possveis de deteco que pode ser acoplado a RP-HPLC para com a determinao das fluoroquinolonas, de seletividade e a deteco espectrofluoromtrica foi superior deteco no ultravioleta em mtodos bioanalticos desempenho satisfatrio sensibilidade (CRDOBA-BORREGO et al., 1999; HUSSAIN et al., 1995; HAN et al., 2005; MASCHER; KIKUTA, 1998; WELL et al., 1998). As fluoroquinolonas, norfloxacino e ciprofloxacino, so estruturas homlogas, classificadas como anfteros que apresentam propriedades semelhantes no tempo de reteno (PILORZ; CHROMA, 2004) que pode ser influenciado pela sua propenso para formar interaes no hidrofbicas com o material de slica gel da fase estacionria (PISTOS et al., 2005).
137
De acordo com Pilorz e Chroma (2004), deve-se ter em mente que a dissociao dos analitos, bem como a troca inica com silanois, so importantes fatores a serem controlados durante a anlise cromatogrfica de fluoroquinolonas.
No presente trabalho foi desenvolvido e validado um mtodo de HPLC-RP com deteco fluoromtrica de norfloxacino em plasma. O mtodo foi aplicado com sucesso no estudo de bioequivalncia de duas formulaes, referncia e teste, de norfloxacino 400 mg em voluntrios sadios. Os cromatogramas livres de analitos apresentaram ausncia de interferentes endgenos da matriz ao longo da janela de eluio para norfloxacino e padro interno (figura 17A). Interferentes potenciais de frmacos que so comumente amoxicilina, administrados ainda no concomitantemente testadas. As com o norfloxacino, como foram condies
cromatogrficas foram escolhidas para uma boa separao de norfloxacino e do padro interno (ciprofloxacino) de picos endgenos no plasma. Com o uso da coluna de alta eficincia Synergi MAXRP C12, com 25% a mais de silanois livres que a C18 convencional, foi possvel obter uma separao satisfatria (fator de resoluo = 1,99) dos picos de norfloxacino e padro interno utilizando uma fase mvel consistida de acetonitrilafosfato de potssio monobsico (pH, 3,0; 25 mM). Os tempos de reteno do norfloxacino e ciprofloxacino foram 4,33 e 4,83 minutos, respectivamente. A resoluo da linha de base foi alcanada em 7 minutos. O perfil cromatogrfico (figura 17B) obtido nas condies estabelecidas foi semelhante ao demonstrado por Vybralov e colaboradores (2005), mas com uma melhor separao dos analitos e com a vantagem do tempo de reteno ser cerca de 3 vezes mais curto, sem necessidade de gradiente de eluio.
138
0.006
Name Retention Time Area
RF10Axl (Ex:300nm, Em:450nm) BRANCO E0106DCD08 005
0.005
0.004
Volts
0.003
0.002 (Ciprofloxacin) 5.0 5.5 (Norfloxacin) 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5
0.001
0.000
0.0
6.0
6.5
7.0
7.5
8.0
Minutes
RF10Axl (Ex:300nm, Em:450nm) CQB Norf loxacino 90 ng/mL em PE E0106DCD08 035
0.05
Name Retention Time Area
0.04
0.01
0.00
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
Ciprofloxacin 4.831 474316 5.0
0.02
0.425 567
Norfloxacin 4.334 41738
Volts
0.03
5.5
6.0
6.5
7.0
7.5
8.0
Minutes
Figura 17. Cromatografia de amostra de plasma branco extrado (A) e de plasma contaminado contendo norfloxacino (90 ng/mL) e padro interno (B).
Comparando nossos resultados com os de Espinosa-Mansilla e colaboradores (2005), que utilizaram um maior volume de plasma (1 mL) que o utilizado no desenvolvimento do mtodo (0,2 mL), descobrimos que o presente mtodo apresenta melhor separao de picos e um menor tempo de reteno.
139
2.2. Linearidade do Mtodo
O mtodo descrito foi validado de acordo com as recomendaes internacionalmente aceitas para mtodos bioanalticos (HUBERT et al., 1999; SHAH et al., 2000).
A curva de calibrao, definida em trs corridas analticas em duplicata, foi linear na faixa de concentrao de 30 a 3500 ng/mL para o norfloxacino em plasma, com todos os coeficientes de correlao maiores que 0,99. A linearidade foi definida pela equao: y = 985,11x 2,52151, onde y representa a relao da altura do pico do norfloxacino e padro interno e x representa a relao da concentrao (Figura 18).
Figura 18. Representao grfica da curva de calibrao y = 985,11x 2,52151, r2= 0,999493.
O limite de quantificao (LQ), definido como o menor ponto da curva de calibrao com preciso e exatido (tabela 10), de grande importncia em estudos farmacocinticos. O LQ do norfloxacino foi de 30 ng/mL. Este limite quase 2% da concentrao mxima esperada (Cmax) do norfloxacino aps uma dose padro (400 mg) na farmacocintica humana e, portanto, apropriado para estudos de biodisponibilidade/bioequivalncia. O LQ obtido no presente estudo foi semelhante ao obtido por Mascher e Kikuta (31 ng/mL), Samanidou e colaboradores (30 ng/mL)
140
e Hussain e colaboradores (25 ng/mL) com volumes de injeo de 20 e 50 l, ou seja, 2 e 5 vezes maior que o aplicado (10 l).
2.3 Preciso e exatido do mtodo
Os testes de preciso do presente mtodo, o qual foi confirmado a recuperao e anlise intra e interlote, foi comparvel com mtodos anteriores (CRDOBA-BORREGO et al., 1999; MYERS; BLUMER, 1987; HUBERT et al., 1999; ABANMI et al., 1996), O percentual do coeficiente de variao representa as medidas de preciso. Os dados de preciso e exatido intra e interlote para o norfloxacino esto resumidos na tabela 6. A validao apresentou uma excelente preciso e exatido para todas as concentraes (30, 90, 1400 e 2800 ng/mL) de norfloxacino. Tabela 6 Dados de preciso e exatido intra e interlote para o norfloxacino na validao em plasma.
Intralote Concentrao Nominal (ng ml ) Mdia DP Exatido (%) Preciso (%) Interlote Mdia DP Exatido (%) Preciso (%) 29,8 2,58 99,5 8,6 93,7 3,91 104,1 4,2 1447,7 41,83 103,4 2,9 2819,8 63,54 100,7 2,3
-1
LQ 30 28,7 0,27 95,8 0,9
CQB 90 90,2 1,35 100,2 1,5
CQM 1400 1450,6 18,29 103,6 1,3
CQA 2800 2873,9 28,99 102,6 1,0
2.4 Recuperao do mtodo
A recuperao das amostras, a qual envolve precipitao de protenas com acetonitrila, foi prxima a 100%. A recuperao mdia relativa do norfloxacino bem como do padro interno (ciprofloxacino) em plasma, determinada com 5 replicatas de cada nvel (90, 1400 e 2800 ng/mL), esto resumidos na tabela 7. A recuperao obtida com o mtodo desenvolvido foi maior que a relatada por Hussain
141
et al. (1995) (87%) ou Mascher e Kikuta (1998) (88%) para norfloxacino na mesma faixa de concentrao. Em seus estudos, o pr-tratamento da amostra envolveu precipitao de protena com cido tricloroactico ou acetonitrila (0,5 mL). O grande volume de acetonitrila (1,6 mL) usado no presente estudo foi provavelmente responsvel (parcialmente, pelo menos) pelo aumento observado na recuperao. Tabela 7 Recuperao do norfloxacino e ciprofloxacino em plasma.
Norfloxacino Concentrao Nominal (ng ml ) 90 1400 2800 Ciprofloxacino Concentrao Nominal (ng ml ) 90 1400 2800 85,6 3,7 1322,9 64,0 2651,8 101,0 88,9 6,3 1311,9 61,7 2688,1 61,0
-1 -1
No-extrado Mdia DP 89,5 2,2 1423,1 84,5 2855,5 96,1 No-extrado Mdia DP
Extrada Mdia DP 92,6 3,1 1424,4 28,6 2918,1 43,3 Extrada Mdia DP
Recuperao (n=15) Mdia (%) 103,5 100,2 102,2 Recuperao (n=15) Mdia (%) 104,4 99,2 101,4
CV (%)
1,9 2,9 1,7 CV (%)
7,4 2,8 1,5
2.5 Estudos de estabilidade do mtodo
A determinao da estabilidade do norfloxacino foi realizada analisando as solues de trabalho, bem como, amostras obtidas imediatamente, horas ou dias subsequentes ao perodo de estoque. Todas as amostras (n=5) foram analisadas usando amostras recm-preparadas da curva de calibrao. Nenhuma alterao na concentrao do norfloxacino foi detectada na soluo de trabalho aps 30 dias de armazenamento com a proteo da luz em 8C. No estudo de estabilidade psprocessamento, nenhuma degradao significativa pode ser observada em amostras resfriadas no autoinjetor por 92 horas. Os resultados mostraram que as reas dos picos de norfloxacino permaneceram praticamente inalteradas e picos extras no foram observados no perodo estudado. Na anlise de 3 conjuntos de amostras de plasma contaminados com concentraes baixa (90 ng/mL), mdia (1400 ng/mL) e alta (2800 ng/mL) de norfloxacino, foram preparados e estocados 18C e submetidos 3 ciclos de congelamento e descongelamento. Os dados
142
demonstraram que as amostras de plasma podem sofrer congelamento e descongelamento pelo menos 3 vezes antes das anlises. Alm disso, tambm foi avaliada a estabilidade de curta durao de amostras de plasma contaminadas, processadas e estocadas em temperatura ambiente (23C) por um perodo de 15 horas. Nenhuma perda significante de norfloxacino aps 15 horas em temperatura ambiente foi observada. Finalmente, para demonstrar a estabilidade de longadurao do norfloxacino, 3 conjuntos de plasma contaminados foram processados e estocados -20C por 44 dias. A anlise das amostras de plasma contaminadas antes e aps estocagem -20C nesse perodo no mostrou nenhuma perda significante do analito (P > 0,05). A mdia e o desvio padro (%) calculados para os diferentes estudos de estabilidade esto demonstrados na tabela 8.
Tabela 8 Teste de estabilidade (teste de estabilidade ps-processamento, estabilidade congelamento e descongelamento, estabilidade de curta (15 horas) e longa durao (44 dias). Nmero de replicatas: n = 5.
Teste de estabilidade ps-processamento (valor em ng/mL) Valor de Valor aps Valor de Valor aps Valor de Valor aps 92h Referncia 92h Referncia 92h Referncia CQB CQM CQA Mdia 90.13 87.58 1418.73 1389.17 2760.08 2736.15 CV (%) 4.4 0.8 2.6 0.4 2.1 1.0 Variao -2.8 -2.1 -0.9 Teste de estabilidade congelamento e descongelamento (valor em ng/mL) Valor de Valor aps Valor de Valor aps Valor de Valor aps 3 Referncia 3 ciclos Referncia 3 ciclos Referncia ciclos CQB CQM CQA 93.953 90.118 1447.215 1350.090 2814.286 2659.866 0.8 1.7 0.8 1.3 2.1 1.2 -4.1 -6.7 -5.5 Teste de estabilidade acima de 15 dias (valor em ng/mL) Valor de Valor aps Valor de Valor aps Valor de Valor aps 15h Referncia 15h Referncia 15h Referncia CQB CQM CQA 97.855 89.550 1409.439 1333.393 2767.454 2626.622 2.7 1.4 3.5 1.0 1.3 0.6 -8.5 -5.4 -5.1 Teste de estabilidade acima de 44 dias (valor em ng/mL) Valor de Valor aps Referncia 44 dias CQB 89.16 78.66 1.2 0.3 -11.8 Valor de Valor aps Referncia 44 dias CQM 1375.96 1225.99 1.7 2.5 -10.9 Valor de Referncia CQA 2742.21 1.0 Valor aps 44 dias 2475.63 1.9 -9.7
Mdia CV (%) Variao
Mdia CV (%) Variao
Mdia CV (%) Variao
143
APLICAO
DO
MTODO ENTRE
BIOANALTICO
EM
ESTUDO
DE
BIOEQUIVALNCIA
DUAS
FORMULAES
CONTENDO
NORFLOXACINO (COMPRIMIDO DE 400MG) EM VOLUNTRIOS SADIOS
O ensaio foi aplicado para avaliar a biodisponibilidade de dois produtos contendo 400 mg de norfloxacino, a fim de determinar se as formulaes teste produzida pela Pharmascience Laboratrios Ltda e referncia produzida pela Merck Sharp & Dohme Farmacutica Ltda no so bioequivalentes em 24 voluntrios. As curvas de concentrao plasmtica mdia da amoxicilina aps administrao oral da formulao teste e referncia versus tempo esto apresentadas na figura 19. As concentraes plasmticas do norfloxacino se mantiveram na faixa padro e acima do limite de quantificao de 30 ng/mL por todo o perodo de quantificao.
2000 1800 1600
Comcentration (ng/mL)
1400 1200 1000 800 600 400 200 0
Referncia Teste
10
15
20
25
Time post-dose (h)
Figura 19. Grfico da curva de concentrao plasmtica (ng/mL) versus tempo (h) de norfloxacino. Representa a mdia das concentraes plasmticas obtidas aps a administrao de duas formulaes de norfloxacino 400 mg em volutrios sadios.
Os parmetros farmacocinticos referentes s formulaes teste e referncia esto descritos na tabela 9. A anlise estatstica da AUC0t, AUC0 e Cmax revelou que no houve bioequivalncia entre as 2 formulaes. A Food and Drug Administration (FDA) props que formulaes farmacuticas sejam
144
consideradas bioequivalentes quando a razo das mdias de Cmax e AUC(0-t) apresentarem-se entre 80125% (considerando um intervalo de confiana de 90%) (FDA, 1985; FDA, 1993). Entretanto, as 2 formulaes no foram consideradas bioequivalentes. Tabela 9 Resultados das anlises de bioequivalncia para relao teste/referncia aps transformao logartmica para os parmetros AUC0-, AUC(0-24h) e Cmax.
Teste/Referncia Anlise Estatstica Parmetro de Anlise Mdia Geomtrica AUC0- ([ng.h]/mL) AUC(0-24h)([ng.h]/mL) Cmax (ng/mL) 91,121 90,497 92,879 Intervalo de confiana 90% (80-125%) (76,33 108,78) (74,59 109,79) (76,43 112,86) No No No Bioequivalncia
CAPTULO 3: Oxcarbazepina
Nas ltimas dcadas vrias frmacos antiepilpticos (Gabapentina, Clobazan, lamotrigina e topiramato), foram introduzidos no mercado, entre os quais a oxcarbazepina (10,11-dihidro-10-oxo-5H-dibenzo-[b, f] azepina-5-carboxamida) um anlogo da carbamazepina, que no se diferenciam em termos de eficcia farmacolgica, porm mostra menos efeitos colaterais indesejados, em comparao com os tradicionais frmacos antiepilpticos (GLAUSER, 2001; CASTILLO et al., 2000; VAN BELLE et al., 1995).
Oxcarbazepina (OXC) considerada eficaz, no s como frmaco de primeira escolha, bem como na terapia coadjuvante da epilepsia parcial (SALLAS et al., 2003; LEVERT et al., 2002a). Atua principalmente na promoo da estabilizao das membranas excitveis, por meio do bloqueio de canais voltagem-dependentes canais de sdio (MAURER et al., 2002; SHORVON, 2000). Em humanos OXC rapidamente metabolizada por enzimas citoslicas no fgado para seu metablito ativo no-txico (10,11-dihidro-10-hidroxicarbamazepina (MHD), que predomina no plasma aps administrao oral, em comparao com OXC (ROUAN et al., 1994;
145
FLESCH, 2004; GONZALEZ-ESQUIVEL et al., 2000; SCHUTZ et al., 1986), e mostram uma farmacocintica linear (MAY et al., 2003; SALLAS et al., 2003). Assim, a identificao, separao e quantificao de OXC e MHD, seu metablito clinicamente relevante, essencial para investigao e monitorizao teraputica de medicamentos biologicamente relacionados, bem como, estudos de bioequivalncia.
No passado, os mtodos mais comumente utilizados para a determinao de OXC e MHD em amostras biolgicas envolviam a cromatografia em fase gasosa (VON UNRUH; PAAR, 1985; VON UNRUH; PAAR, 1986). Atualmente, a cromatografia lquida de alta eficincia acoplada deteco ultravioleta (VAN BELLE et al., 1995; LEVERT et al., 2002a; ROUAN et al., 1994; GONZALEZ-ESQUIVEL et al., 2000; MILES et al., 2004; LEVERT et al., 2002b) tem sido amplamente utilizada para a quantificao simultnea de OXC e MHD em fluidos biolgicos. Contudo, a limitao comumente discutida de todos estes mtodos de determinao de frmacos antiepilpticos o fato da existncia da grande similaridade (propriedades fsico-qumicas e estruturas) existente entre estes compostos (LEVERT et al., 2002a). Por outro lado, geralmente no comum a descrio da determinao simultnea de OXC e MHD em plasma humano utilizando CLAE seguido de separao seletiva por deteco de espectrometria de massa. Dois mtodos foram descritos para a quantificao simultnea de OXC e seu metablito ativo (MHD) em plasma humano por cromatografia lquida acoplada a espectrometria de massa com ionizao qumica por presso atmosfrica (MAURER et al., 2002) ou ionizao por electrospray (BRETON et al., 2005) com um limite de quantificao (LQ) de 100 ng/ml e 300 e 400 ng/ml para OXC e MHD, respectivamente.
Desenvolveu-se um mtodo seguro, rpido, sensvel e seletivo utilizando LC-MS/MS para quantificao simultnea de OXC e MHD no plasma humano com menor LQ (20 e 40 ng/ml para OXC e MHD, respectivamente), que combina menor volume de amostra de plasma, curto tempo de reteno e um simples preparo de amostra, com preciso e exatido adequadas.
146
1 ETAPA CLNICA
O estudo foi delineado, de forma a permitir que se obtenham os parmetros farmacocinticos relevantes, visando a averiguao do perfil farmacocintico. No caso em estudo, tais parmetros foram obtidos diretamente a partir da determinao da concentrao plasmtica de OXC e MHD, baseado na aplicao de um modelo no-compartimental prprio para avaliao destas concentraes, aps a administrao do medicamento por via oral.
Por conseguinte, a finalidade primria da Etapa Clnica foi a coleta de amostras de sangue dos voluntrios para medir (na Etapa Analtica) nveis plasmticos das substncias aps sua administrao oral.
O desenho consistiu de um estudo aberto, sem replicao, de 8 voluntrios sendo 4 homens e 4 mulheres no grvidas. O termo voluntrio normal ou sadio deve ser interpretado como indivduo adulto, sem anormalidades clnicolaboratoriais capazes de prejudicar a interpretao do experimento ou aumentar a sensibilidade do indivduo ao potencial txico do frmaco em estudo. A idade variou entre 21 e 46 anos (22,75 8,91 anos), altura variando de 157 a 179 cm (147,38 0,09 cm) e ndice de massa corporal mdio de 20,25 2,86 kg/m2.
Os voluntrios qualificados para participar do estudo foram internados por 01 perodo de aproximadamente 24 horas. Amostras sangneas para a determinao dos nveis plasmticos dos frmacos foram obtidas antes da administrao (tempo zero e pool) e aps a administrao, seguindo o seguinte esquema:
OXC e MHD 30, 60, 90, 120, 150, 180, 210, 240, 270, 300, 330, 360, 390, 420, 450, 480, 600, 720, 1440 e 2880 minutos.
147
2 DESENVOLVIMENTO E VALIDAO DO MTODO BIOANALTICO PARA QUANTIFICAO DA OXCARBAZEPINA
2.1 Otimizao dos procedimentos gerais
No presente trabalho foi desenvolvido e validado um mtodo para quantificao de OXC e MHD em plasma. As condies cromatogrficas e parmetros de espectrometria de massa foram otimizadas tal que a resultante de cromatogramas MRM extrados apresentaram todas as reas integradas e relaes sinais-rudos consistentes (S/N > 15) para OXC, MHD e padro interno. Usando uma coluna analtica Phenomenex Luna C18 5m (150mm4,6 mm), foi possvel obter uma resoluo de picos satisfatria para OXC, MHD e padro interno utilizando uma fase mvel constituda de acetonitrila-gua (50:50 v/v) + 20mM cido actico.
As estruturas qumicas bem como o espectro de massa (on precursor e fragmento) dos analitos e padro interno esto apresentados na figura 20. Vrios ons fragmento foram observados no produto dos ons para os analitos e padro interno. Os principais ons fragmentos em m/z 208, m/z 194 e m/z 204 foram escolhidos na aquisio do MRM para OXC, MHD e padro interno, respectivamente.
148
Figura 20. Full-scan dos espectros dos ons fragmento de [M+H]+ e as estruturas da (a) OXC, (b) MHD e (c) d10-carbamazepina.
149
Cromatogramas MRM de amostra de plasma branco e contaminado com OXC, MHD e padro interno submetidos ao procedimento extrao lquido-lquido esto representados na figura 21A e 21B, respectivamente. Um cromatograma de uma amostra de voluntrio aps administrao de OXC suspenso oral (6%) est representado na figura 21C. Sob as condies do mtodo proposto, nenhuma interferncia (efeito matriz) foi observada de compostos endgenos aps a extrao de amostras de plasma contaminadas ao longo do tempo de eluio e encontraramse tempos de reteno de 2,44 0,3, 1,87 0,3 e 2,89 0,2 minutos para OXC, MHD e padro interno, respectivamente. No se observou efeito de cross-talk no tempo de reteno dos compostos, demonstrado, dessa forma, boa seletividade.
150
Figura 21. Cromatogramas MRM: (a) uma amostra de plasma em branco, (b) uma amostra de plasma branco contaminada com OXC (20 ng / ml) e MHD (40 ng / ml), (c) um voluntrio amostra 1,5 h aps a administrao oral de OXC suspenso (6%) em voluntrios saudveis. Picos I-III referem-se ao MHD, OXC e PI, respectivamente.
151
Foram publicados dois mtodos para quantificao simultnea de OXC e seu metablito ativo (MHD) em plasma por cromatografia lquida acoplada a espectrometria de massa (MAURER et al., 2002; BRETON et al., 2005). O primeiro mtodo (MAURER et al., 2002) foi concludo com xito, porm envolve um prvio tratamento das amostras biolgicas e eludo por gradiente, de modo que o tempo total de anlise (a partir do pr-tratamento at a anlise no equipamento) maior, existindo outras desvantagens, tais como: (1) maior volume de plasma (0,2 mL) em relao ao presente mtodo (0,1 ml). Um menor volume de amostra especialmente vantajoso quando em estudos farmacocinticos em crianas; (2) a corrida analtica cerca de duas vezes maior (em torno de 6 minutos) do que a verificada no presente mtodo (3,5 minutos). Comparando com o segundo mtodo (VOLOSOV et al., 1999), observa-se que tambm h necessidade de utilizao de gradiente de eluio, maior volume de amostra (0,3 ml) e um tempo de reteno maior para OXC (em torno de 18 minutos) e MHD (em torno de 12 minutos), em comparao com aqueles encontrados no presente estudo para OXC (2,44 minutos) e MHD (1,87 minutos). Foi utilizada no estudo uma simples etapa no mtodo de extrao com ter-diclorometano antes da corrida analtica, realizada com fase mvel isocrtica. Deve-se considerar que um rpido processo de extrao um fato importante para anlise de rotina. Alm disso, sabido que um mtodo isocrtico geralmente preferido por causa da sua convenincia, simplicidade e reprodutibilidade. Assim, o presente mtodo tem suficiente seletividade e pode ser aplicado com sucesso para a quantificao da OXC e MHD em amostras de plasma. O procedimento relativamente simples de preparao das amostras e um curto tempo de anlise cromatogrfica apresentada permitem um grande nmero de quantificaes de amostras (150 a 200 amostras por dia).
2.2 Linearidade do mtodo
A qualidade dos dados bioanalticos dependente da boa obteno e interpretao da curva de calibrao. O teste de homoscedasticidade um passo importante a ser tomado no estudo da curva de calibrao para verificar varincias dos dados (ALMEIDA et al., 2002). Neste estudo, o teste de homoscedasticidade foi realizado plotando resduo versus concentrao obtida no ensaio intralote por HPLC-
152
MS/MS e mediante aplicao de um teste-F (CARDONE et al., 1990). Uma vez que os dados apresentaram varincia no-uniforme, o mtodo de regresso com diferentes pesos (1/x, 1/x2, 1/y e 1/y2) foi usado para escolher o menor percentual do erro relativo da curva de calibrao. De acordo com estas circunstncias, o fator escolhido foi 1/x2 (tabela 10). Assim, as curvas de calibrao foram definidas pela equao: y = 0,00568 + 0,00296x5,708x2 para OXC e y = 0,00749 + 0,00178x5,708x2 para MHD, onde y a relao das reas dos picos do analito/padro interno e x a concentrao (ng/ml) do analito. Todos os coeficientes de correlao (r2) foram prximos a 1 (0,9986 a 0,9994). Tabela 10 Preciso e exatido intralote das curvas de calibrao.
Concentrao plasmtica (ng/ml)
OXC 20 750 1500 2250 3000 3750 4500 5250 19,99 0,26 768,82 16,46 1558,17 25,50 2115,04 62,20 3040,24 198,16 3400,62 74,78 4659,85 120,48 5469,95 51,35 1,31 2,14 1,64 2,94 6,52 2,20 2,59 0,94 -0,05 2,51 3,88 -6,00 1,34 -9,32 3,55 4,19 0,1 39,98 1,90 1529,95 18,82 3086,33 56,46 4260,31 231,44 6111,70 298,49 6999,86 220,81 9123,15 477,68 10935,65 200,52 4,76 1,23 1,83 5,43 4,88 3,15 5,24 1,83 -0,05 2,00 2,88 -5,33 1,86 -6,67 1,37 4,15 0,2
Concentrao calculada (MdiaDP)(ng/ml)
RSD (%) (n=3)
Intralote RE (%)
Soma RE(%)
MHD 40 1500 3000 4500 6000 7500 9000 10500
Soma RE (%)
153
2.3 Preciso e exatido do mtodo Os dados para preciso e exatido intra e interlote para OXC e MHD esto resumidos na tabela 11. Os erros relativos dos valores variam entre 1,7 e 4,5% e o desvio padro relativo (% R.S.D.) de 0,15 a 7,1% para ambos compostos e as amostras dos controles de qualidade apresentaram bom ajuste individual para a linha de regresso aplicada. Tabela 11 Preciso e exatido na quantificao de OXC e MHD em amostras de plasma (anlise de amostras de plasma contaminado em quatro concentraes diferentes).
Intralote Concentrao Plasma (ng/ml) OXC 20 60 2000 4000 MHD 40 120 4000 8000 38,04 2,34 119,5 4,48 3919 67,34 7487,4 318,68 6,2 3,7 1,7 4,3 4,9 0,41 2,0 6,4 38,586 2,01 119,7 5,30 4006,5 137,04 7736,6 350,89 5,2 4,4 3,4 4,5 3,5 0,25 0,15 3,29 19,86 0,86 64,3 1,56 2050,9 43,54 3804,6 114,4 4,3 2,4 2,1 3,0 0,7 7,1 2,5 4,9 20,11 0,24 63,1 3,44 2050,3 84,12 3880,2 282,29 1,2 4,2 2,9 2,3 0,5 5,2 2,5 3,0 Concentrao calculada (MdiaDP) (ng/ml) RSD (%) (n=8) RE(%) Interlote Concentrao calculada (MdiaDP) (ng/ml) RSD (%) (n=8) RE(%)
O limite de quantificao baseado no R.S.D. (%) e RE (%) foi 20 e 40 ng/ml para OXC e MHD, respectivamente. Este limite foi prximo a 1% da concentrao mxima (Cmax) de OXC e 2% para MHD, sendo suficiente para realizao dos estudos de biodisponibilidade. Comparando com os valores de LQ obtidos por mtodos anteriormente publicados utilizando cromatografia por ionizao qumica (MAURER et al., 2002) ou espectrometria de massa por electrospray (BRETON et al., 2005), revela que o presente mtodo cerca de 5 e 20 vezes mais sensvel. Valores menores de LQ (1 ng/ml) foram registrados em estudos envolvendo quantificao de OXC e MHD por LC-MS/MS em amostras de
154
microdialisis (LANCKMANS et al., 2006), porm deve-se ter em mente que essas amostras esto livres de endgenos e protenas, no sendo necessrio o preparo das amostras para anlise. Pienimaki e colaboradores (PIENIMAKI et al., 1995) publicaram um mtodo por HPLC-UV que atingiram valores de LQ (55 e 24 ng/ml para OXC e MHD, respectivamente) prximo ao do presente estudo, porm exige um volume maior de amostra (500 l) com dispendioso e trabalhoso tempo de preparo. Calculou-se tambm a sensibilidade terica do mtodo proposto, para OXC e MHD, estimando que com uma amostra de 100 l, com reconstituio em 100 l de acetonitrila, volume de injeo de 5 l e LQ de 20 ng/ml para OXC e 40 ng/ml para MHD encontrou-se 100 pg e 200 pg, respectivamente. Isto representa um considervel aumento da sensibilidade, em comparao com outros mtodos similares. Assim, estes aspectos refletem algumas vantagens do presente mtodo sobre os demais publicados anteriormente.
2.4 Recuperao do mtodo
As recuperaes relativas de OXC e MHD determinadas em trs concentraes diferentes (CQB, CQM e CQA) foram 105,4, 89,2 e 92,8%, e 88,4, 88,7 e 90,6%, respectivamente, sendo a mdia geral da recuperao de 95,8% para OXC e 86,7% para MHD. Valores de recuperaes (117% para OXC e 81% para MHD) foram registrados por Breton et al. (2005) no intervalo de concentrao de 500 a 20000 ng/ml, porm, como j mencionado, utilizando amostras de plasma com maior volume de amostra (0,3 ml). No intervalo de concentrao de 10 a 35 ng/ml e utilizando uma amostra de plasma (0,2 mL) com pr-tratamento muito mais complexo que o do presente estudo, Maurer e colaboradores (2002) relataram recuperaes de 59,6 a 79,2% para OXC e 57,4 a 63.9% para MHD. Assim, o mtodo proposto apresentou uma melhor recuperao para amostras de plasma atravs da aplicao de um simples processo de extrao e com menor tamanho de amostra (0,1 ml).
155
2.5 Estudos de estabilidade do mtodo
A anlise da estabilidade da OXC e MHD foi realizada analisando as solues de trabalho, bem como, amostras imediatamente, horas ou dias subsequentes do perodo de estoque. Todas as amostras (n=5) foram analisadas usando amostras recm-preparadas da curva de calibrao. Nenhuma alterao na concentrao da OXC e MHD foi detectada na soluo de trabalho aps 1 ms de armazenamento com a proteo da luz em 8C. No estudo de estabilidade psprocessamento, nenhuma degradao significativa pode ser observada em amostras resfriadas (8C) no autoinjetor por 92 horas. Os resultados mostraram que as reas dos picos de OXC e MHD permaneceram praticamente inalteradas e picos extras no foram observados no perodo estudado. Na anlise de 3 conjuntos de amostras de plasma contaminados (CQB, CQM, CQA) de OXC e MHD, foram preparados e estocados 18C e submetidos a 3 ciclos de congelamento e descongelamento. Os dados demonstraram que as amostras de plasma podem sofrer congelamento e descongelamento pelo menos 3 vezes antes das anlises. Alm disso, tambm foi avaliada a estabilidade curta-durao de amostras de plasma contaminada processadas e estocadas em temperatura ambiente (23C) por um perodo de 15 horas. Nenhuma perda significante de norfloxacino aps 15 horas em temperatura ambiente foi observada. Finalmente, para demonstrar a estabilidade de longadurao do norfloxacino, as amostras foram processadas e estocadas -20C por 30 dias. A anlise das amostras de plasma contaminadas antes e aps estocagem -20C nesse perodo no mostrou nenhuma perda significante do analito (P > 0,05). Os erros relativos (RE%) calculados para os diferentes estudos de estabilidade esto demonstrados na tabela 12.
156
Tabela 12 Estabilidade das amostras de controle de qualidade da OXC e MHD em plasma sob diferentes condies de estoque (n=5). OXC (ng/ml) 60 0h 137h 0 ciclos 3 ciclos 0h 7h 0 dias 30 dias 3,6 0,8 2,6 1,7 4,2 3,3 1,2 2,7 2000 1,0 3,7 3,6 2,2 2,2 2,7 2,2 1,0 4000 5,2 3,9 3,3 3,3 5,7 7,4 2,0 2,8 120 6,6 5,1 3,6 5,2 2,4 1,2 3,5 1,2 MDH (ng/ml) 4000 9,1 5,9 2,8 2,6 2,5 2,1 2,8 1,0 8000 8,4 6,2 2,7 2,2 3,1 3,2 1,6 1,0
Teste de estabilidade ps-processamento (RE%)
Teste de congelamento e descongelamento (RE%)
Teste de estabilidade curta-durao (RE%)
Teste de estabilidade de longa
3 APLICAO DO MTODO BIOANALTICO EM ESTUDO FARMACOCINTICO DE UMA FORMULAO CONTENDO OXCARBAZEPINA (SUSPENSO 6%) EM VOLUNTRIOS SADIOS
Aps validao, o mtodo proposto foi aplicado em um estudo piloto para anlise de amostras de plasma retiradas de oito voluntrios sadios imediatamente antes e aps 30, 60, 90, 120, 150, 180, 210, 240, 270, 300, 330, 360, 390, 420, 450, 480, 600, 720, 1440 e 2880 minutos da administrao oral de uma dose de 10 ml de OXC suspenso (6%). O perfil da curva de concentrao plasmtica mdia da OXC e MHD versus tempo est apresentado na figura 22. As concentraes plasmticas de OXC e MHD estavam na faixa de concentrao normal e reais, acima da quantificao limite para todo o perodo de amostragem.
157
Figura 22. Perfil da curva de concentrao plasmtica mdia de OXC e MHD versus tempo aps administrao de 10 ml de uma suspenso oral de oxcarbazepina (6%) em voluntrio sadios (n=8).
158
CONSIDERAES FINAIS E CONCLUSO
159
CONSIDERAES FINAIS
Considerando os dados obtidos, pode-se concluir que os mtodos analticos propostos oferecem a oportunidade para obter parmetros farmacocinticos com uma adequada preciso e exatido, pois esto dentro dos limites necessrios exigidos pela legislao para os estudos bioequivalncia e farmacocinticos. Alm disso, os ensaios so rpidos e o tempo da corrida analtica no ultrapassou os 7 minutos para os trs estudos.
A capacidade de manterem-se inalterados em pequenas variaes nos parmetros caracterizou a robustez dos mtodos, possibilitando a intercambialidade entre laboratrios e doseamento em outros fluidos biolgicos. Possuem vantagens em termos de custos, em comparao com outros mtodos descritos, pois utilizam procedimentos de preparo de amostras relativamente simples sendo desnecessria a utilizao de pr-tratamento em cartuchos e pr-colunas. Nas condies cromatogrficas descritas, as substncias analticas foram bem separadas dos padres interno com alta sensibilidade e reprodutibilidade.
As limitaes da pesquisa so possveis influncias que podem ou no ser controladas pelo pesquisador. Este tipo de abordagem est relacionado ao emprego de recursos e tcnicas estatsticas que visem quantificar os dados coletados. No desenvolvimento da pesquisa de natureza quantitativa devem-se formular hipteses e classificar a relao entre as variveis para garantir a preciso dos resultados, evitando contradies no processo de anlise e interpretao. Assim, foi observada relativa dificuldade quanto gerao, interpretao e anlise dos resultados estatsticos uma vez que esses parmetros farmacocinticos foram processados por empresa (Pharmacience Laboratrios Ltda) especializada e autorizada pela Agncia Nacional de Vigilncia Sanitria.
Os mtodos analticos mostraram-se precisos, pois os coeficientes de variao para os controles de qualidade, baixo, mdio e alto permaneceram abaixo de 15% e abaixo de 20% para o controle de qualidade com a mesma concentrao
160
do limite de quantificao. Verificou-se acurcia, pois os valores mdios permaneceram dentro dos 15% do valor nominal para o controle de qualidade baixo, mdio e alto e no se desviou mais que 20% de limite de quantificao.
Os mtodos foram considerados sensveis, pois os menores limites de quantificao dos mtodos (LQ=0,5 g/mL; 30ng/mL; 20/40 ng/mL, para amoxicilina, norfloxacino e OXC/MHD, respectivamente) apresentaram um coeficiente de variao inferior a 20%.
Os mtodos mostraram-se especficos j que no houve interferncia nos tempos de reteno dos picos dos analitos superior a 20% da resposta do limite de quantificao.
A estabilidade dos mtodos foi assegurada, atravs dos testes de congelamento e descongelamento, onde se verificou no haver degradao devido temperatura, em 3 ciclos de congelamento e descongelamento, bem como no psprocessamento, curta e longa durao de estocagem.
Os
mtodos
analticos
utilizados
HPLC/RP
LC-MS-MS
para
quantificao de norfloxacino e OXC/MHD em plasma humano, desenvolvido e validado, apresentam vantagens sobre os j descritos. A quantificao de amoxicilina demonstrou resultados semelhantes aos j publicados, embora utilizando outras tcnicas de deteco como espectrometria de massa.
CONCLUSO
Os trs mtodos bioanalticos
propostos foram desenvolvidos,
devidamente validados e quantificam com exatido (acurcia) e preciso (reprodutibilidade) os parmetros farmacocinticos de formas farmacuticas orais de amoxicilina, norfloxacino e oxcarbazepina em plasma de voluntrios sadios, de modo que so vlidos para o emprego em estudos de bioequivalncia que envolvam esses frmacos.
161
REFERNCIAS
162
REFERNCIAS ABIFAM. Perfil mercadolgico dos medicamentos genricos. [S.l.], 2001. ALMEIDA, A .M.; CASTEL-BRANCO, M. M.; FALCO, A. C. J. Linear regression for calibration lines revisited: weighting schemes for bioanalytical methods. Chromatogr. B, v. 774, p. 215, 2002. ANVISA (Brasil). RDC n 10, de 2 de janeiro de 2001. Regulamento tcnico para medicamentos genricos. Dirio Oficial da Repblica Federativa do Brasil, Poder executivo, Braslia, DF, 09 de janeiro de 2001. ANVISA (Brasil). Resoluo RE n 899, de 29 de maio 2003. Guia para validao de mtodos analticos e bioanalticos. Dirio Oficial da Repblica Federativa do Brasil, Poder executivo, Braslia, DF, 02 de juho de 2003. ANDERSON, G. D.; LEVY, R. H. Phenobarbital: chemistry and biotransformation. In: LEVY, R. H.; MATTSON, R. H.; MELDRUM, B. S. (Ed.). Antiepileptic drugs. 4th ed. New York: Raven Press, 1995. p. 371-377 ANTIGNAC, J. P.; BIZEC, B. L.; MONTEAU, F.; POULAIN, F.; ANDRE, F. Collisioninduced dissociation of corticosteroids in electrospray tandem mass spectrometry and development of a screening method by high performance liquid chromatography/tandem mass spectrometry, RCMS, v. 14, p. 33-39, 2000. ASSOCIAO GRUPO DE ANALISTAS DE RESDUOS DE PESTICIDAS (GARP). Manual de Resduos de Pesticidas em Alimentos. [S.l.], 1999. Apostila. ATKINSON, H. C.; BEGG, E. J.; DARLOW, B. A. Drug in human milk: clinical pharmacokinetic considerations. Clin. Pharmacokinet., v. 14, p. 217-240, 1988. ATKINSON, J. A.; DANIELS, C. E.; DECRICK, R. L.; GRUDZINSKAS, C. V.; MARKEY, S. P. Principles of clinical pharmacology. London: Academic Press, London, 2001. AUGUSTO, F.; ANDRADE, J. C.; CUSTDIO, R. The linear range of a calibration curve. Chemkeys, p. 1-8, 2000. BAGLIE S.; ROSALEN, P. L.; FRANCO, L. M.; RUENIS, A. P.; BAGLIE, R. C.; FRANCO, G. C.; SILVA, P.; GROPPO, F. C. Comparative bioavailability of 875 mg amoxicillin tablets in healthy human volunteers. Int. J. Clin. Pharmacol. Therap., v. 43, p. 350-354, 2005. BALL, A. P.; DAVEY, P.G.; GEDDES, A.M.; FARREI, I.D.; BROOKES, G. Clavulanic acid and amoxycillin: a clinical, bacteriological, and pharmacological study. Lancet, p. 620-623, 1980. BARR, W.; ZOLA, E. M.; CANDLER, E. L. Differential absorption of amoxicilin from the human small and large intestine. Clin. Pharmacol. Ther., v. 56, p. 279-285, 1994.
163
BARROS NETO, B.; PIMENTEL, M. F.; ARAJO, M. C. U. Recomendaes para calibrao em qumica analtica - Parte I. Fundamentos e calibrao com um componente (calibrao univariada). Quim. Nova, v.25, n.5, p.856-865, 2002. BENBROOK, D. M.; MILLER, R. V. Effects of norfloxacin on DNA metabolism in Ps. Aeruginosa. Antimicrob. Agents Chemother., v. 29, p. 1-6, 1986. BENET, L. Z. Effect of rout of administration and distribution on drug action. J Pharmacokinet. Biopharm., v. 6, p. 559-585, 1978. BENET, L. Z. Pharmacokinetic parameters: wich are necessary to define a drug substance? Eur. J. Res. Dis., v. 65, suppl. 134, p. 45-61, 1984. BENET, L. Z.; GALEAZZI, R. L. Noncompartimental determination of the steady-state volume of distribution. J. Pharm. Sci., v. 68, p. 1971-1974, 1979. BERG, R. G.; MURTA, A. L. M.; KUGLER, W. O mtodo das adies padro aplicado anlise cromatogrfica quantitativa de fenis em guas residuais. Quim. Nova, v. 11, p. 288-291, 1988. BERGERON, M. G.; THABET, M.; ROY, R.; LESSARD, C.; FOUCAULT, P. Norfloxacin penetration into human renal and prostatic tissues. Antimicrob. Agents Chemother., v. 26, p. 110-111, 1984. BIALER, M. Oxcarbazepine - chemistry, biotransformation and pharmacokinetics. In: LEVY, E. H.; MATTSON, R.; MELDRUM, B. S. et al. (Ed.). Antiepileptic Drugs. Philadelphia: Lippincott-Williams and Wilkins, 2002. cap. 45, p. 459-465. BLUMBERG, P. M.; STROMINGER, J. L. Interaction of penicillin with the bacterial cell: penicillin-binding proteins and penicillin-sensitive enzymes. Bacteriol. Rev., v. 38, p. 291, 1974. BOX, G. E. P.; HUNTER, W. G.; HUNTER, J. S. Statistics for Experimenters. New York: Wiley, 1987. BRIDGES, J. W. Metabolism and molecular interactions related toxicity. In: (Fowler, B.A. (Ed.). Mechanism of cell injury: Implications for human heath. New York: John Wiley & Sons, 1987. BRETON, H.; COCIGLIO, M.; BRESSOLLE, F.; PEYRIERE, H.; BLAYAC, J. P.; HILLAIRE-BUYS, D. Liquid chromatographyelectrospray mass spectrometry determination of carbamazepine, oxcarbazepine and eight of their metabolites in human plasma. J. Chromatogr. B, v. 828, p. 80-90, 2005. BRODIE, P. Physicochemical factors in drug absorption. In: BINNS, T. B. (Ed.). Absorption and distribution of drug. Baltimore: The Williams & Wikins Co., 1964.
164
BRUCE, P.; MINKKINEN, P.; RIEKKOLA, M. L. Practical method validation: validation sufficient for an analysis method. Mikrochim. Acta, v. 128, p. 93-106, 1998. BULL, J. P. The historical development of clinical therapeutics trials. J. Chron. Dis., v. 10, p. 218-248, 1959. BURNS, D. T.; DANZER, K.; TOWNSHEND, A. Use of the term recovery and apparent recovery in analytical procedures. Pure Appl. Chem., v. 74, p. 22012205, 2002. CAUSON, R. Validation of chromatographic methods in biomedical analysis. Viewpoint and discussion. J. Chromatograp. B, v. 689, p. 175-180, 1997. CARDONE, M. J.; WILLAVIZE, S. A.; LACY, M. E. Method validation revisited: a chemometric approach. Pharm. Res., v. 7, p. 154-160, 1990. CASTILLO, S.; SCHMIDT, D. B.; WHITE, S. Oxcarbazepine add-on for drug-resistant partial epilepsy. Cochrane Database Syst. Rev., v. 3, CD002028, 2000. CHARLES, B.; CHULAVATNATOL, S. Simple analysis of amoxicillin in plasma by high performance liquid chromatography with internal standardization and ultraviolet detection. Biomed. Chromatogr, v. 7, p. 204-207, 1993. CHASIN, A. M.; CHASIN, M.; SALVADORI, M. C. Validao de mtodos cromatogrficos em anlises toxicolgicas. Rev. Farm. Bioquim. USP, v. 30, p. 4146, 1994. CHUI, Q. S. H.; ZUCCHINI, R. R.; LICHTIG, J. Qualidade de medies em qumica analtica. Estudo de caso: determinao de cdmio por espectrofotometria de absoro atmica com chama. Quim. Nova, v. 24, p. 374-380, 2001. CODEX ALIMENTARIUS. Commission on Methods of Analysis and Sampling; Criteria for Evaluating Acceptable Methods of Analysis for Codex, 1995. COFSKY, R. D.; DUBONCHET, L.; LENDESMAN, S. H. Recovery of norfloxacin in feces after administration of a single oral dose to human volunteers. Antimicrob. Agents Chemother., v. 26, p. 110-111, 1984. COLEBROOK, L.; PURDIE, A. W. Treatment of 106 cases of puerperal fever by sulphanilamide. Lancet, p. 1237-1242, 1291-1294, 1937. COLLINS, C. H.; BRAGA, G. L.; BONATO, P. S. Introduo a mtodos cromatogrficos. 7. ed. Campinas, SP: UNICAMP, 1997. CRDOBA-BORREGO, M.; CRDOBA-DAZ, M.; CRDOBA-DAZ, D. Validation of a high-performance liquid chromatographic method for the determination of norfloxacin and its application to stability studies (photo-stability study of norfloxacin). J. Pharm. Biomed. Anal., v. 18, p. 919926, 1999.
165
COZZARELLI, N. R. DNA gyrase and the supercoiling of DNA. Science, v. 207, p. 953-960, 1980. CRUMPLIN, G. C.; KENWRIGHT, M.; HIRST, T. Investigations into the mechanism action of the antibacterial agent norfloxacin. J. Antimicrob. Chemother., v. 13, p. 923, 1984. CUADROS-RODRGUEZ, L.; GMIZ-GRACIA, L.; ALMANSA-LPEZ, E. M.; BOSQUE-SENDRA, J. M. Calibration in chemical measurement processes. II. A methodological approach, Trends Anal. Chem., v. 20, p. 620-635, 2001. CUADROS-RODRGUEZ, L.; GARCIA-CAMPAA, A. M.; ALMANSA-LPEZ, E. M.; EGEA-GONZLEZ, F. J.; CANO, M. L. C.; FRENICH, A. G.; MARTINEZ-VIDAL, J. L. Correction function on biased results due to matrix effects: Application to the routine analysis of pesticide residues. Anal. Chim. Acta, v. 478, p. 281-301, 2003. CULLMANN, W.; DICK, W. A simple enzyme assay for the simultaneous determination of penicillin derivatives and clavulanic acid in biological fluids. Immun. Infect., v. 14, p. 188-190, 1986. CUSTODIO, R.; DE ANDRADE, J. C.; AUGUSTO, F. O ajuste de funes matemticas a dados experimentais. Quim. Nova, v. 20, p. 219-225, 1997. DE ANDRADE, J. C. O papel dos erros determinados em anlises qumicas. Quim. Nova, v. 10, p. 159-165, 1987. DOUSA, M.; HOSMANOVA, R. Rapid determination of amoxicillin in premixes by HPLC. J. Pharm. Biomed. Anal., v. 37, p. 373, 2005. ERAH, P. O.; GODDARD, A. F.; BARRETT, D. A.; SHAW, P. N.; SPILLER, R. C. J. The stability of amoxycillin, clarithromycin and metronidazole in gastric juice relevance to the treatment of Helicobacter pylori infection. Antimicrob. Agents Chemother., v. 39, p. 5-12, 1997. EGEA-GONZLEZ, F. J.; TORRES, M. E. H.; LPEZ, E. A.; CUADROSRODRGUEZ, L.; VIDAL, J. L. M. Matrix-effects of vegetable commodities in electroncapture detection applied to pesticide multiresidue analysis. J. Chromatogr. A, v. 966, p. 155, 2002. ENGLE, E. C.; MANES, S. H.; DRLICA, K. Differential effects of antibiotics inhibiting gyrase. J. Bacteriol., v. 149, p. 92-98, 1983. ESPINOSA-MANSILLA, A.; DE LA PEA, A. M.; GMEZ, D. G.; SALINAS, F. HPLC determination of enoxacin, ciprofloxacin, norfloxacin and ofloxacin with photoinduced fluorometric (PIF) detection and multiemission scanning: application to urine and serum. J. Chromatogr. B, v. 822, p. 185193, 2005. EURACHEM WORKING GROUP. The Fitness for Purpose of Analytical Methods, A Laboratory Guide to Method Validation and Related Topics, 1998.
166
FERGUSSON, F. R.; DAVEY, A. F.; TOPLEY, W. W. C. The value of mixed vaccines in the prevention of the common cold. J. Hyg., v. 26, p. 98-109, 1927. FINK, G. D. Drug abuse. In: BRODY, T. M.; LARNER, J.; MINNEMAN, K. P. (Ed.). Human pharmacology: molecular to clinical. 3 ed. St. Louis, MO: Mosby, 1998. FLESCH, G. Overview of the Clinical Pharmacokinetics of Oxcarbazepine. Clin. J. Invest., v. 24, p. 185, 2004. FOOD AND DRUG ADMINISTRATION. Bioavailability requirements. Fed. Regist., n. 320, p. 154-173, 1985. and bioequivalence
FOOD AND DRUG ADMINISTRATION. Bioavailability and bioequivalence requirements; abbreviated applications; proposed revisions-FDA. Proposed rule. Fed. Regist., v. 63, n. 223, p. 64222-64228, 1998a. FOOD AND DRUG ADMINSTRATION. Guidance for industry, bioanalytical methods validation for human studies. ., 1998b. Disponvel em:<http://www.fda.gov/cder/guidance/1221.pdf>. Acesso em: 21 Dec. 2010. FOOD AND DRUG ADMINSTRATION. In vivo Pharmacopeial Forum, v. 19, p. 6501-6508, 1993. bioequivalence guidances.
GLAUSER, T.A. Oxcarbazepine in the treatment of epilepsy. Pharmacotherapy, v. 21, p. 904-919, 2001. GOLDSTEIN, A.; ARONOW, L.; KALMAN, S.M. Principles of drug action: The basis of pharmacology, 2 ed. John Wiley & Sons Inc., New York, 1974. GONZALEZ-ESQUIVEL, D.F.; ORTEGA-GAVILN, M.; ALCANTARA-LOPEZ, G. JUNG-COOK, H. Plasma level monitoring of oxcarbazepine in epileptic patients. Arch. Med. Res., v. 31, p. 202-205, 2000. GOODMAN & GILMAN. As bases farmacolgicas da teraputica, 11 ed., Guanabara Koogan, Rio de Janeiro, 2007. GORDON, R.C.; GAMEY, C.; KIRBY, W.M.M. Comparative clinical pharmacology of amoxicillin and ampicillin administered orally. Antimicr Ag Chemother, v. 1, p. 504507, 1972. GORDON, R.C.; WINSHELL, E.B. Pharmacological studies of 6 [D(-)-amino-phydroxyphenylacetamidol] penicilanic acid in humans. Antimicr Ag Chemoter, v. 1, p. 423-426, 1970. GLAUSER, T.A. Oxcarbazepine in the treatment of epilepsy. Pharmacotherapy v. 21, p. 904-919, 2001. GRANT, S.M.; FAULDS, D. Oxcarbazepine: a review of its pharmacology and therapeutic potential in epilepsy, trigeminal neuralgia and affective disorders. Drugs, v. 43, p. 873-888, 1992.
167
GREEN, J.M. A practical guide to analytical method validation. Anal. Chem., v. 68, p. A305-309, 1996. GREENWOOD, M.; YULE, G.U. The statistics of anti-typhoid and anti-cholera inoculations and interpretation of such statistics in general. Proc R Soc Med, v. 8, p.113-194, 1915. GUYTON, A.C. Tratado de Fisiologia Mdica. 4 ed., Guanabara Koogan, Rio de Janeiro, 1973. HAN, Y.; WU, X.; YANG, J.; et al. The fluorescence characteristic of the yttriumnorfloxacin system and its analytical application. J Pharm Biomed Anal. v. 38, p. 528531, 2005. HANDFIELD, H.H.; CLARK, H.; WALLACE, J.F.; HOLMES, K.K.; TURCK, M. Amoxicilin, a new penicillin antibiotic. Antimicr Ag Chem, v. 3, p. 262-265, 1973. HENION, J.; BREWER, E.; RULE, G. Sample preparation for LC/MS/MS: analyzing biological and environmental samples. Anal Chem, v. 70, p. 650-656, 1998. HEYDEN, Y.V. Analysis. v. 22, p. M27, 1994. HILL, A.R.C; REYNOLDS, S.L. Guidelines for in-house validation of analytical methods for pesticide residues in food and animal feeds. Analyst. v. 124, p. 953, 1999. HOIZEY G., LAMIABLE D., FRANCES C., TRENQUE T., KALTENBACH M., DENIS J. E MILLART H. Simultaneous determination of amoxicillin and clavulanic acid in human plasma by HPLC with UV detection. Journal of Pharmaceutical and Biomedical Analysis v. 30, p. 661-666, 2002. HOLMES, O.W. Currents and counter currents in medical science. In Work, v. 9, p. 185, 1891. HOOPER, D.C.; WOLFSON, J.S. Mode of action of the quinolone antimicrobial agents. Review of recent information. Reviews of Infectious Diseases, v. 11, p. S902- S911, 1989. HOOPER, W.D.; DICKINSON, R.G.; DUNSTAN, P.R. Oxcarbazepine: preliminary Clinical and pharmacokinetic studies on a new anticonvulsant. Clin Exp Neurol. v. 24, p. 105-112. HORWITZ, W. Protocol for the design, conduct and interpretation of met hod-perf ormance studies. Pure Appl. Chem. v. 67, p. 331-343, 1995. HORWITZ, W.; KAMPS, L.R.; BOYER, K.W. J. Assoc. Offic. Anal. Chem. v. 63, p. 1344, 1980.
168
HOUTKOOPER, M.A.; LAMMERTSMA, A.; MEYER, J.W. Oxcarbazepine (GP 47.680): a possible alternative to carbamazepine? Epilepsia, v. 28, p. 693-698, 1987. HUBER, L. Validation of analytical methods: review and strategy. LC-GC Int., v. 11, p. 96-105, 1998. HUBERT P.; CHIAP, P.; CROMMEN J. The SFSTP guide on the validation of chromatographic methods for drug bioanalysis: from the Washington Conference to the laboratory. Anal Chim Acta. v. 391, p. 135148, 1999. HUSSAIN, M.S.; CHUKWUMAEZE-OBIAJUNWA, V.; MICETICH, R.G. Sensitive high performance liquid chromatographic assay for norfloxacin utilizing fluorescence detection. J Chromatogr B. v. 63, p. 379384, 1995. INSTITUTO NACIONAL DE METROLOGIA, NORMALIZAO E QUALIDADE INDUSTRIAL (INMETRO). Vocabulrio Internacional de Termos Fundamentais e Gerais de Metrologia, 2 ed., 2000. INSTITUTO NACIONAL DE METROLOGIA. Normalizao e Qualidade Industrial (INMETRO); Orientaes sobre Validao de Mtodos de Ensaios Qumicos, DOQCGCRE-008, 2003. INTERNACIONAL STANDARD ORGANIZATION. Precision of Test Methods, ISO 5725, 1994. INTERNATIONAL CONFERENCE ON HARMONISATION (ICH); Validation of Analytical Procedures: Definitions and Terminology, Q2A (CPMP/ICH/381/95), 1995. INTERNATIONAL CONFERENCE ON HARMONISATION (ICH); Validation of Analytical Procedures: Methodology, Q2B (CPMP/ICH/281/95), 1995. INTERNATIONAL STANDARD ORGANIZATION. General Requirements for the Competence of Testing and Calibration Laboratories, ISO/IEC 17025, 1999. INTERNATIONAL STANDARD ORGANIZATION. Statistics-Vocabulary Symbols- Part 1: Probability and General Statistical Terms, ISO 3534-1, 1993. and
JENKE, D.R. Chromatographic Method Validation: A Review of Current Practices and Procedures. Instrument. Sci. Technol. v. 25, p. 345-359, 1997. JENKE, D.R. Chromatographic Method Validation: A Review of Current Practices and Procedures. Part II. Guidelines for Primary Validation Parameters. Instrument. Sci. Technol. v. 26, p. 1-18, 1998a. JENKE, D.R. Chromatographic Method Validation: A Review of Current Practices and Procedures. Part II. Guidelines for Primary Validation Parameters. Instrument. Sci. Technol. v. 26, p. 19-35, 1998b.
169
JUERGENS, U. HPLC analysis of antiepileptic drugs in blood samples: microbore separation of fourteen compounds. J Liquid Chromatogr v. 10, p. 507-532, 1987. KLOTZ, U.; AVANT, G.R.; HOUYUMPA, A.; SCHENKER, S.; WILKINSON, G.R. The effects of age and liver descase on the disposition and elimination of diazepam in adult man. J Clin Invest, v. 55, p. 347-359, 1975. KOSMIDIS, P.; WILLIANS, J.D.; ANDREWS, J. Amoxicillin pharmacology, bacteriology and clinical studies. Br J Clin Pract, v. 26, p. 341-346, 1972. KRAUWINKEL, W.J.; VOLKERS-KAMERMANS, N.J.; VAN ZIJTVELD, J. Determination of amoxicillin in human plasma by high-performance liquid chromatography and solid phase extraction. J Chromatogr, v. 617, p. 334-338, 1993. KRISTENSEN, O.; KLITGAARD, N.A.; JONSSON, B.; et al. Pharmacokinetics of 10OH-carbazepine, the main metabolite of the antiepileptic oxcarbazepine, from serum and saliva concentrations. Acta Neurol Scand, v. 68, p. 145-150, 1983. KRULL, I.; SWARTZ, M.; Frequently Asked Questions about. Analytical Method Validation. LC-GC. v. 16, p. 464-467, 1998. KYUANG-HWAN YOON; SO-YOUNG LEE; WON KIM; JONG-SEI PARK; HIE-JOON KIM., Simultaneous determination of amoxicillin and clavulanic acid in human plasma by HPLC-ESI mass spectrometry. J. Chromatogr. B, v. 813, p. 121-127, 2004. LANCKMANS, K.; CLINCKERS, R.; VAN EECKHAUT, A.; SARRE, S.; SMOLDERS, I.; MICHOTTE, Y. Use of microbore LC-MS/MS for the quantification of oxcarbazepine and its active metabolite in rat brain microdialysis samples. J. Chromatogr. B. v. 831, p. 205, 2006. LANAS, F.M. Cromatografia em Fase Gasosa, Acta: So Carlos, 1993. LEENHEER, A.P.; NELIS, H.J.; LAMBERT, W.E.; BAUWENS, R.M. Chromatography of fat-soluble vitamins in clinical chemistry. J. Chromatogr. v. 429, p. 3-58, 1988. LEITE, F. Validao em Anlise Qumica, 4a ed., Editora tomo: Campinas, 2002. LEVERT, H.; ODOU, P.; ROBERT, H. LC determination of oxcarbazepine and its active metabolite in human serum. J Pharm Biomed Anal, v. 28, p. 517-25, 2002a. LEVERT, H.; ODOU, P.; ROBERT, H. Simultaneous determination of four antiepileptic drugs in serum by highperformance liquid chromatographyBiomed. Chromatogr. v. 16, p. 19-24, 2002b. LIND, J. A treatise of the scurvy. Edinburgh: Sands Murray & Cochran (eds), 1753.
170
LLOYD, P.; FLESCH, G.; DIETERLE, W. Clinical pharmacology pharmacokinetics of oxcarbazepine. Epilepsia, v. 35, p. S10-13, 1994.
and
LOBO, N.C. Desenvolvimento De Um Mtodo Analtico de Norfloxacina em Plasma de Voluntrios Sadios Usando Cromatografia Lquida de Alta Eficincia no Modo Reverso. Dissertao de Mestrado, Universidade So Francisco Bragana Paulista SP, p. 8-39, 2001. LOUIS, P.C.A. Essay on clinical instruction. London: S. Highley (ed), 1834. MASCHER, H.J.; KIKUTA, C. Determination of norfloxacin in human plasma and urine by high-performance liquid chromatography and fluorescence detection. J Chromatogr A. v. 812, p. 381385, 1998. MASSART, D.L.; SMEYERS-VERBEKE, J.; VANDEGINSTE, B. An introduction to method validation. Analysis. v. 22, p. M14-16, 1994. MATAR, K.M.; NICHOLLS, P.J.; AL-HASSAN, M.I.; et al. A rapid micromethod for simultaneous measurement of oxcarbazepine and its active metabolite in plasma by high-performance liquid chromatography. J Clin Pharm Ther. v. 20, 229-234, 1995. MATUSZEWSKI, B.K.; CONSTANZER, M.L.; CHAVEZ-ENG, C.M. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal Chem. v. 75, p. 3019, 2003. MAURER, H.H. Liquid chromatography-mass spectrometry in forensic and clinical toxicology. J Chromatogr B, v. 713, p. 3-25, 1998. MAURER, H.H.; KRATZSCH, C.; WEBER, A.A.; PETERS, F.T.; KRAEMER, T. Validated assay for quantification of oxcarbazepine and its active dihydrometabolite 10-hydroxycarbazepine in plasma by atmospheric pressure chemical ionization liquid chromatography/mass spectrometry. J. Mass Spectrom. v. 37, p. 687-692, 2002. MAY, T.W.; KORN-MERKER, E.; RAMBECK, B. Clinical pharmacokinetics of oxcarbazepine. Clin Pharmacokinet. v. 42, p. 1023-1042, 2003. MCLEAN, M.J.; SCHMUTZ, M., WAMIL, A.W. Oxcarbazepine: mechanisms of action. Epilepsia. v. 35, p. S5-9, 1994. MENELAOU A.; SOMOGYI A.A.; BARCLAY M.L.; BOCHNER F. Simultaneous quantification of amoxycillin and metronidazole in plasma using high-performance liquid chromatography with photodiode array detection. Journal of Chromatography B. v. 731, p. 261-266, 1999. MENGE, G.; DUBOIS, J.P. Determination of OXC in human plasma by highperformance liquid chromatography. J Chromatogr. v. 275, p. 189-194, 1983.
171
MENGE, G.; DUBOIS, J.P.; BAUER, G. Simultaneous determination of carbamazepine, oxcarbazepine and their main metabolites in plasma by liquid chromatography. J Chromatogr. v. 414, 477-483, 1987. MILES, M.V.; TANG, P.H.; RYAN, M.A.; GRIM, S.A.; FAKHOURY, T.A.; STRAWSBURG, R.H.; DEGRAUW, T.J.; BAUMANN, R.J. Feasibility and limitation of oxcarbazepine monitoring using salivary monohydroxycarbamazepine (MHD). Ther. Drug Monit., v. 26, p. 300-304, 2004. MILLER, J.C.; MILLER, J.N. Statistics for Analytical Chemistry, 2 ed., Ellis Horwood: Chichester, 1988. MIYAZAKI, K.; OHTANI, K.; SUNADA, K.; ARITA, T. Determination of ampicillin, amoxicillin, cephalexin and cephradine in plasma by high-performance liquid chromatography using fluorimetric detection. J Chromatogr, v. 276, p. 478-82, 1983. MUTH, P.; METZ, R.; BECK, H. BOLTEN, W.W.; VERGIN, H. Improved highperformance liquid chromatographic determination of amoxicillin in human plasma by means of column switching. J Chromatogr A, v. 729, p. 259-266, 1996. MYERS, C.M.; BLUMER, J.L. High-performance liquid chromatography of ciprofloxacin and its metabolites in serum, urine and sputum. J Chromatogr. v. 422, p. 153164, 1987. NASIR M.I.; AHMAD A.; NAJI M.N. Bioequivalence evaluation of two brands of amoxicillin/clavulanic acid 250/125 mg combination tablets in healthy human volunteers: Use of replicate design approach. Biopharmaceutics & Drug Disposition. v. 25, p. 367-372, 2004. NEBERT, D.W.; GONZALEZ, F.J. P450 genes: structure, evolution and regulation. Annu Rev Biochem, v. 56, p. 945-993, 1987. NEDELMAN, J.R.; HOSSAIN, M.; CHANG, S.W.; et al. Oxcarbazepine: analysis of concentration-efficacy/safety relationships [abstract]. Neurology. v. 52, p. A524, 1999. NEU, H.C. Antimicrobial activity and human pharmacology of amoxicillin. J Infect Dis, v. 129, p. S123-S131, 1974. NEU, H.C.; WINSHELL, E.B. Pharmacological studies of 6 [D(-)-amino-phydroxyphenylacetamidol] penicillanic acid in humans. Antimicr Ag Chemoter, v. 1, p. 423-26, 1970. NORRBY, S.R. Pharmacokinetics of norfloxacin: clinical implications. European Journal of Chemotherapy and Antibiotics, v. 3, p. 19-25, 1983. PAGE, C.; CURTIS, M.; SUTTER, M.; WALKER, M.; HOFFMAN, B. Integrated pharmacology. Mosby, Edinburgh, 2002.
172
PAINTAUD, G.; ALVN, G.; DAHL, M.L.; GRAHNN, A.; SJVALL, J.; SVENSON, J.O. Nonlinearity of amoxicilin absorption kinetics in human. Eur Clin Pharmacol, v. 43, p. 283-288, 1992. PATSALOS, P.N.; ELYAS, A.A.; ZAKRZEWSKA, J.M. Protein binding of oxcarbazepine and its primary active metabolite, 10-hydroxy Clin Pharmacokinet. v. 42, p. 12, 2003. PATSALOS, P.N.; ELYAS, A.A.; ZAKRZEWSKA, J.M. Protein binding of oxcarbazepine and its primary metabolite, l0-hydroxycarbazepine, in patients with trigeminal neuralgia. Eur J Clin Pharmacol. v. 39, p. 413-415, 1990. PIENIMAKI, P.; FUCHS, S.; ISOJARVI, J.; VAHAKANGAS, K. Improved detection and determination of carbamazepine and oxcarbazepine and their metabolites by high-performance liquid chromatography. J. Chromatogr. B. v. 673, p. 97-105, 1995. PILORZ, K.; CHROMA, I. Isocratic reversed-phase high-performance liquid chromatographic separation of tetracyclines and flumequine controlled by a chaotropic effect. J Chromatogr A. v. 1031, p. 303305, 2004. PISTOS, C.; TSANTILI-KAKOULIDOU, A.; KOUPPARIS, M. Investigation of the retention/pH profile of zwitterionic fluoroquinolones in reversed-phase and ioninteraction high performance liquid chromatography. J Pharm Biomed Anal. v. 39, p. 438443, 2005. PRESCOTT, L.F.; NIMMO, W.S. Drug absorption. ADIS Press, New York, 1981. RIBANI, M.; BOTTOLI, C.B.G.; COLLINS, C.H.; JARDIM, I.C.S.F.; MELO, L.F.C. Validao em mtodos cromatogrficos e eletroforticos. Qumica Nova. v. 27, p. 771-780, 2004. RIBEIRO, A.N.F. Medicamentos Genricos. Informaes para farmacuticos ou profissionais da sade. Maio, 2000. RIDOUT, G.; SANTUS, G.C.; GUY, R.H. Pharmacokinetic considerations in the use of new transdermal formulations. Clinical Pharmacokinet, v. 15, p. 114-131, 1988. ROBERTS, M.S.; DONALDSON, J.D.; ROWLAND, M. Models of hepatic elimination: Comparison of stochastic models to describe residence time distributions and to predict the influence of drug distribution, enzimeheterogeniety, and systemic recycling on hepatic elimination. J Pharmacokinetic Biopharm, v. 16, p. 41-83, 1988. ROWLAND, M. TOZER, T.N. Clinical Pharmacokinetics. Concepts and Applications. 3ed. Lea & Febiger. London, 1995. VAN BELLE, K.; VERFAILLIE, I.; EBINGER, G.; MICHOTTE, Y. Liquid chromatographic assay using a microcolumn coupled to a U-shaped optical cell for
173
high-sensitivity ultraviolet absorbance detection of oxcarbazepine and its major metabolite in microdialysates. J. Chromatogr. B. v. 672, p. 97-102, 1995. VAN ROOYEN, G.F.; BADENHORST, D.; SWART, K.J.; HUNDT, H.K.L.; SCANES,T.; HUNDT, A.F. Determination of carbamazepine and carbamazepine 10,11-epoxide in human plasma by tandem liquid chromatographymass spectrometry with electrospray ionisation. J. Chromatogr. B. v. 769, p. 1-7, 2002. ROUAN, M.C.; LECAILLON, J.B.; GODBILLON, J.; et al. The effect of renal impairment on the pharmacokinetics of oxcarbazepine and its metabolites. Eur J Clin Pharmacol. v. 47, p. 161-167, 1994. SALLAS, W.M.; Milosavljev, S.; D'Souza, J.; et al. Pharmacokinetic drug interactions in children taking oxcarbazepine. Clin Pharmacol Ther. v. 74, p. 138149, 2003. SAMANIDOU V.F.; DEMETRIOU C.E.; PAPADOYANNIS I.N. Direct determination of four fluoroquinolones, enoxacin, norfloxacin, ofloxacin, and ciprofloxacin, in pharmaceuticals and blood serum by HPLC. Anal Bioanal Chem. v. 375, p. 623 629, 2003. SCHIITZ, H.; FELDMANN, K.F.; FAIGLE, J.W. The metabolism of 14Coxcarbazepine in man. Xenobiotica. v. 16, p. 769-778, 1986. SETH, S.D.; BEOTRA, A.; SETH S. Comparative bioavailability of two brands of norfloxacin. J Assoc Physicians India. v. 43, p. 324326, 1995. SHABIR, G.A. Validation of high-performance liquid chromatography methods for pharmaceutical analysis: Understanding the differences and similarities between validation requirements of the US Food and Drug Administration, the US Pharmacopeia and the International Conference on Harmonization. J. Chromatogr. A. v. 987, p. 57-66, 2003. SHAH, V.P.; MIDHA, K.K.; FINDLAY, J.W. Bioanalytical method validationa revisit with a decade of progress. Pharm Res. v. 17, p. 15511557, 2000. SHERVINGTON, L.A.; ABBA, M.; HUSSAIN, B.; et al. The simultaneous separation and determination of five quinolone antibiotics using isocratic reversed-phase HPLC: application to stability studies on an ofloxacin tablet formulation. J Pharm Biomed Anal. v. 39, p. 769775, 2005. SHORGET, L.; ANDREW, B.C. Pharmacokinetics, 6 ed. Norwalk, Connecticut, 1941. SHORVON, S. Oxcarbazepine: a review. Seizure, v. 9, p. 75-9, 2000. SCHUTZ, H.F.; FELDMANN, K.F.; FAIGLE, J.W.; KRIEMLER, H.; WINKLER, T. The metabolism of 14C-oxcarbazepine in man. Xenobiotica, v. 16, p. 769-778, 1986.
174
SIMON, C.; STILLE, W. Antibiotikatherapie in Klinik und Praxis. Schattauer, Stuttgart, New York, p. 48-50, 1985. SINGER, S.J.; NICHOLSON, G.L. The fluid-mosaic model of the structure of membrane. Science, v. 175, p. 720-731, 1972. SIUZDAK, G. The expanding role of mass spectrometry in biotechnology. Journal of American Society for Mass Spectrometry, v. 15, p. 625, 2004. SNYDER, L.R.; KIRKLAND, J.J.; GLAJCH, J.L. Practical HPLC Method Development, 2a ed., Wiley: New York, cap. 15, 1997. SOURGENS, H.; STEIN BREDE, H.; VERSCHOOR, J.S.;BERTOLA, M.A.; RAYER, B. Bioequivalence study of a novel Solutab tablet formulation of amoxicillin/clavulanic acid versus the originator film-coated tablet. Int J Clin Pharmacol Ther, v. 39, p. 7582, 2001. SKOOG, D.A.; LEARY, J.L. Principles of instrumental analysis, 4 ed., Harcourt Brace College Publishers, Saunders College Publishing: New York, 1992. SUTHERLAND, R.; ROLINSON, G.N. Amino-p-hydroxybenzil penicillin (BRL 2333), a new semi synthetic penicillin: in vitro evaluation. Antimicr Ag Chemother, v. 1, p. 411, 1970. SUTTON, H.G. Cases of rheumatic fever treated for the most part by mint water. Guys Hosp Rep, v. 2, p. 292, 1865. SWANSON, B.N.; BOPPANA, V.K.; VLASSES, P.H.; ROTMENSCH, H.H.; FERGUSON, R.K. Norfloxacin disposition after sequentially increasing oral doses. Antimicrobial Agents and Chemotherapy. v. 23, p. 284-288, 1983. SWARTZ, M.E.; KRULL, I. S. Validao de mtodos cromatogrficos. Pharm. Technol. v. 2, p. 12-20, 1998. THOMPSON, M.; ELLISON, S. L. R.; WOOD, R.; Harmonized Guidelines for Single Laboratory Validation of Methods of Analysis. Pure Appl. Chem. v. 74, p. 835-855, 2002. THOMPSON, M.; ELLISON, S.L.R.; FAJGELJ, A.; WILLETTS, P.; WOOD, R.; Harmonised guidelines for the use of recovery information in analytical measurement. Pure Appl. Chem. v. 71, p. 337-348, 1999. TOMASZ, A. From penicillin-binding proteins to the lysis and death of bacteria. Rev Infect Dis, v. 1, p. 434, 1979a. TOMASZ, A. The mechanism of irreversible antimicrobial effect of penicillins: how the beta lactam antibiotics kill and lyse bacteria. Ann Rev Microbiol, v. 33, p. 113, 1979b.
175
UNITED STATES FOOD AND DRUG ADMINISTRATION (US-FDA); Guidance for Industry, Analytical Procedures and Methods Validation, 2000. UNITED STATES FOOD AND DRUG ADMINISTRATION (US-FDA); Guidance for Industry, Bioanalytical Method Validation, 2001. UNITED STATES FOOD AND DRUG ADMINISTRATION (US-FDA); Reviewer Guidance, Validation of Chromatographic Methods, 1994. UNITED STATES PHARMACOPEIA CONVENTION. US Pharmacopeia 24, Validation of Compendia Methods <1225>, Rockville, 1999. VAN DER VOET, H.; VAN RHIJN, J.A.H.; VAN DE WIEL, H.J. Inter-laboratory, time, and fitness-for-purpose aspects of effective validation. Anal. Chim. Acta. v. 391, p. 159-171, 1999. VESSMAN, J.; STEFAN, R.I.; STADEN, J.F.V.; DANZER, K.; LINDNER, W.; BURNS, D.T.; FAJGELJ, A.; MLLER, H. Selectivity in analytical chemistry. Pure Appl. Chem. v. 73, p. 1381-1386, 2001. VIAL, J.; JARDY, A. Interlaboratory studies: the best way to estimate the characteristics of dispersion of an HPLC method and a powerful tool for analytical transfers. Chromatographia. v. 53, p. S-141-148, 2001. VOLOSOV, A.; XIAODONG, S.; PERUCCA, E.; YAGEN, B.; SINTOV, A.; BIALER, M. Enantioselective pharmacokinetics of 10-hydroxycarbazepine after oral administration of oxcarbazepine to healthy Chinese subjects. Clin. Pharmacol. Ther. v. 66, p. 547-553, 1999. VON UNRUH, G.E.; PAAR, W.D. Gas chromatographic assay for oxcarbazepine and its main metabolites in plasma. J Chromatogr, v. 345, p. 67-76, 1985. VON UNRUH, G.E.; PAAR, W.D. Gas chromatographic/mass spectrometric assays for oxcarbazepine and its main metabolites, 10-hydroxy-carbazepine and carbaze-pine-10,11-trans-diol. Biomed Environ Mass Spectrom, v. 13, p. 651-656, 1986. VYBRALOV, Z.; NOBILIS, M.; ZOULOVA, J.; et al. High-performance liquid chromatographic determination of ciprofloxacin in plasma samples. J Pharm Biomed Anal. v. 37, p. 851858, 2005. WAXMAN, D.J.; STROMINGER, J.L. Penicillin-binding proteins and the mechanisms of action of beta-lactam antibiotics. Annu Rev Biochem, v. 52, p. 825, 1983. WELL, M.; NABER, K.G.; KINZIG-SCHIPPERS, M.; et al. Urinary bactericidal activity and pharmacokinetics of enoxacin versus norfloxacin and ciprofloxacin in healthy volunteers after a single oral dose. Int J Antimicrob Agents. v. 10, p. 3138, 1998. WIBAMA, J.I.; FOWKES, D.; SHAW, P.N.; BARRET, D.A. Measurement of amoxicillin in plasma and gastric samples using high-performance liquid
176
chromatography with fluorimetric detection. J Chromatogr B, v. 774, p. 141-148, 2002. WISE, R. Norfloxacin a review of pharmacology and tissue penetration. Journal of Antimicrobial Agents and Chemotherapy. v. 13, p. 59-64, 1984. WOLFSON, J.S.; HOOPER, D.C. The fluoroquinolones: structures, mechanisms of action and resistance, and spectra of activity in vitro. Antimicrobial Agents and Chemotherapy. v. 28, p. 581-586, 1985. WORLD HEALTH ORGANIZATION. Expert Committee on Specifications for Pharmaceutical Preparations; 32 report, WHO Technical Report Series, n.823, Geneva, 1992. YOON K.H.; LEE S.Y.; KIM W.; PARK J.S.; KIM H.J. Simultaneous determination of amoxicillin and clavulanic acid in human plasma by HPLCESI mass spectrometry. Journal of Chromatography B. v. 813, p. 121-127, 2004. YUAN Z.; RUSSILIE H.Q.; CANAFAX D.M. High-performance liquid chromatographic analysis of amoxicillin in human and chinchilla plasma, middle ear fluid and whole blood. Journal of Chromatography B. v. 9, p. 361-366, 1997. YUAN, Z.; RUSSLIE, H.Q.; CANAFAX, D.M. Sensitive assay for measuring amoxicillin in human plasma and middle ear fluid using solid-phase extraction and reversed-phase high-performance liquid chromatography. J Chromatogr B, v. 674, p. 93-99, 1995.
177
GLOSSRIO
178
GLOSSRIO Biodisponibilidade Absoluta (F): a frao do frmaco sistemicamente absorvida e a disponibilidade do frmaco da forma farmacutica quando comparada com a biodisponibilidade do frmaco aps administrao intravenosa. usualmente calculada como a razo da rea sob a curva (ASC) da concentrao plasmtica versus tempo, para a forma farmacutica administrada oralmente em relao ASC obtida aps administrao intravenosa do frmaco. Um valor F de 0,80 ou 80% indica que apenas 80% do frmaco ficou disponvel sistemicamente da forma farmacutica. Biodisponibilidade Oral: indica a velocidade e a extenso de absoro de um princpio ativo em uma forma de dosagem, a partir de sua curva concentrao versus tempo na circulao sistmica ou a partir de sua excreo urinria. a frao da dose de um medicamento, oralmente administrado, que foi absorvida e atinge a circulao como princpio ativo (Lei n 9.787, de 10/2/99). Biodisponibilidade Relativa: disponibilidade sistmica do frmaco a partir de sua forma farmacutica quando comparada a uma formulao de referncia. calculada como a razo da rea sob a curva da concentrao plasmtica versus tempo, ASC para a forma farmacutica em relao ao ASC do referncia. Uma biodisponibilidade relativa de F = 1,0 ou 100% implica que a biodisponibilidade a partir da forma farmacutica a mesma, mas no indica a completa absoro sistmica do frmaco. A determinao da biodisponibilidade muito importante em estudos de medicamentos. Denominao Comum Brasileira: denominao do frmaco ou princpio
farmacologicamente ativo aprovado pelo rgo federal responsvel pela vigilncia sanitria (Lei n 9.787, de 10/2/99). Denominao Comum Internacional: denominao do frmaco ou princpio farmacologicamente ativo recomendada pela Organizao Mundial de Sade (Lei n 9.787, de 10/2/99).
179
Bioequivalncia: dois medicamentos so ditos bioequivalentes quando so administrados na mesma dose molar, nas mesmas condies experimentais e no apresentam diferenas estatisticamente significativas em relao biodisponibilidade (BRASIL, 2003a). Droga: a matria prima mineral, vegetal ou animal da qual se podem extrair um ou mais princpios ativos. De acordo com esta acepo, os agentes teraputicos de origem sinttica no so drogas. Frmaco: a substncia qumica de constituio definida que pode ter aplicao em Farmcia, seja como preventivo, seja como curativo, seja como agente de diagnstico; a ser aceita esta definio, a matria prima mineral, vegetal ou animal da qual se podem extrair uma ou mais bases medicamentosas no frmaco, pois sua constituio qumica no necessariamente conhecida. Medicamento: produto farmacutico, tecnicamente obtido ou elaborado, com finalidade profiltica, curativa, paliativa ou para fins de diagnstico". (Lei n 5.991, de 17/12/73). uma forma farmacutica terminada que contm o frmaco, geralmente em associao com adjuvantes farmacotcnicos. Medicamentos Bioequivalentes: so medicamentos equivalentes farmacuticos que, ao serem administrados na mesma dose molar, nas mesmas condies experimentais, no apresentam diferenas estatisticamente significativas em relao biodisponibilidade, segundo a Resoluo RDC n 135, de 29 de maio de 2003. Medicamento Referncia: medicamento inovador registrado no rgo federal responsvel pela vigilncia sanitria e comercializado no Pas, cuja eficcia, segurana e qualidade foram comprovadas cientificamente junto ao rgo federal competente, por ocasio do registro (Lei n 9.787, de 10/2/99). Medicamento Genrico: medicamento similar a um produto de referncia ou inovador, que se pretende ser com este intercambivel, geralmente produzido aps a expirao ou renncia da proteo patentria ou de outros direitos de
180
exclusividade, comprovada a sua eficcia, segurana e qualidade, e designado pela DCB ou, na sua ausncia, pela DCI (Lei n 9.787, de 10/2/99). Estudo Aberto: tanto o pesquisador responsvel como o sujeito da pesquisa so informados e tm conhecimento sobre a medicao que ser administrada. Estudo Cruzado: em uma das fases da pesquisa administra-se ao sujeito da pesquisa (voluntrio) o medicamento referncia e na outra o medicamento teste, ou viceversa. O mesmo voluntrio deve ser submetido s duas formulaes. Analito: composto qumico especfico a ser mensurado, podendo ser o frmaco no-transformado, biomolcula ou seu derivado, metablito ou produto de degradao em uma matriz biolgica (BRASIL, 2003c). Matriz Biolgica: material distinto de origem biolgica, que pode ser amostrado e processado de modo reprodutvel (BRASIL, 2003c). Controle de Qualidade: amostra de matriz biolgica adicionada do analito, usada para monitorar o desempenho de um mtodo bioanaltico e para avaliar a integridade e validade dos resultados das amostras desconhecidas, analisadas numa corrida individual (BRASIL, 2003c). Corrida Analtica: conjunto completo de amostras em estudo, com um nmero apropriado de padres e controles de qualidade para sua validao e que tem sua anlise completa nas mesmas condies (BRASIL, 2003c). Padro Interno: composto, geralmente, com caractersticas estruturais similares ao analito, adicionado aos padres de calibrao e amostras em concentraes conhecidas e constantes, para facilitar a determinao do analito (BRASIL, 2003c). Padro de Calibrao: matriz biolgica a qual foi adicionada uma quantidade conhecida de analito. Os padres de calibrao so usados para construir a curva de calibrao, com a qual so determinadas as concentraes do analito nos controles de qualidade e nas amostras desconhecidas em estudo (BRASIL, 2003c).
181
Preciso: representa o grau de repetibilidade entre os resultados de anlises individuais, quando o procedimento aplicado diversas vezes numa mesma amostra homognea, em idnticas condies de ensaio (BRASIL, 2003c). Exatido: representa o grau de concordncia entre os resultados individuais encontrados e um valor aceito como referncia (BRASIL, 2003c). Linearidade: corresponde capacidade do mtodo de fornecer resultados diretamente proporcionais concentrao da substncia em exame (analito) (BRASIL, 2003c). Limite de Deteco: menor concentrao de um analito que o procedimento bioanaltico consegue diferenciar confiavelmente do rudo de fundo (BRASIL, 2003c). Limite Inferior de Quantificao: menor quantidade de um analito numa amostra que pode ser determinada quantitativamente com preciso e exatido aceitveis (BRASIL, 2003c). Especificidade: habilidade do mtodo bioanaltico de medir e diferenciar o analito de componentes que possam estar presentes na amostra, tais como metablitos, impurezas, compostos de degradao ou componentes da matriz (BRASIL, 2003c). Reprodutibilidade: preciso entre dois laboratrios. Tambm representa a preciso do mtodo sob as mesmas condies operacionais, num curto perodo de tempo (BRASIL, 2003c). Estabilidade: parmetros que visa determinar se um analito mantm-se quimicamente inalterado numa dada matriz sob condies especficas, em determinados intervalos de tempo (BRASIL, 2003c). Recuperao: eficincia de extrao de um mtodo analtico, expressa como a porcentagem da quantidade conhecida de um analito, obtida da comparao dos
182
resultados analticos de amostras branco acrescidas de padro e submetidas ao processo de extrao, com os resultados analticos de soluo padro no extradas (BRASIL, 2003c). rea sob a Curva da Concentrao Plasmtica versus Tempo, de Zero ao Infinito ASC0- : relaciona-se com a quantidade ou a extenso de absoro do frmaco. A quantidade da absoro sistmica do frmaco diretamente relacionada com ASC que geralmente calculado pelo mtodo trapezoidal; onde ASC[0- ] = ASC[0-t] + Ct/K, e expresso em unidades de concentrao vezes tempo. Concentrao Mxima Atingida no Plasma - Cmax: concentrao mais elevada do frmaco atingida na circulao sangnea aps administrao de um frmaco. Relaciona-se a intensidade da resposta farmacolgica. O Cmax ideal deve estar dentro da janela teraputica. Constante de Eliminao K: representa a soma de todas as taxas constantes para remoo do frmaco do corpo, incluindo a taxa constante da excreo renal e metabolismo (biotransformao) como descrito atravs da equao: K = K + Km, onde K = constante de excreo renal e Km = constante de metabolismo. Determina a taxa de eliminao do frmaco dos vasos sangneos. Serve para determinar a concentrao do frmaco em um tempo t. inversamente relacionada meia vida do frmaco: K= 0,693/t1/2. Meia Vida de Eliminao do Frmaco t1/2: representa o tempo em que a concentrao do frmaco no plasma reduzida a metade. Calcula-se atravs do logaritmo neperiano (ln) de 2 (ln2= 0,693) dividido pela constante de eliminao. t1/2 = ln2/K. Tempo correspondente Concentrao Mxima Atingida no Plasma - Tmax: tempo correspondente para que o frmaco atinja a concentrao mxima (Cmax).
183
ANEXOS
184
ANEXO A LISTAS DE RANDOMIZAO PARA AMOXICILINA
N 1. 2. 3. 4. 5. 6. 7. 8. 9.
PERODO I Amoxicilina (cpsula AMOXIL Amoxicilina (cpsula Amoxicilina (cpsula AMOXIL Amoxicilina (cpsula AMOXIL Amoxicilina (cpsula Amoxicilina (cpsula AMOXIL AMOXIL AMOXIL Amoxicilina (cpsula AMOXIL Amoxicilina (cpsula AMOXIL Amoxicilina (cpsula Amoxicilina (cpsula AMOXIL Amoxicilina (cpsula AMOXIL AMOXIL Amoxicilina (cpsula AMOXIL
PERODO II AMOXIL Amoxicilina (cpsula AMOXIL AMOXIL Amoxicilina (cpsula AMOXIL Amoxicilina (cpsula AMOXIL AMOXIL Amoxicilina (cpsula Amoxicilina (cpsula Amoxicilina (cpsula AMOXIL Amoxicilina (cpsula AMOXIL Amoxicilina (cpsula AMOXIL AMOXIL Amoxicilina (cpsula AMOXIL Amoxicilina (cpsula Amoxicilina (cpsula AMOXIL Amoxicilina (cpsula
10. 11. 12. 13. 14. 15. 16. 17. 18. 19. 20. 21. 22. 23. 24.
185
ANEXO B LISTAS DE RANDOMIZAO PARA NORFLOXACINO N 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. 17. 18. 19. 20. 21. 22. 23. 24. PERODO I Floxacin Norfloxacino Floxacin Norfloxacino Floxacin Norfloxacino Floxacin Norfloxacino Floxacin Norfloxacino Floxacin Norfloxacino Floxacin Norfloxacino Floxacin Norfloxacino Floxacin Norfloxacino Floxacin Norfloxacino Floxacin Norfloxacino Floxacin Norfloxacino PERODO II Norfloxacino Floxacin Norfloxacino Floxacin Norfloxacino Floxacin Norfloxacino Floxacin Norfloxacino Floxacin Norfloxacino Floxacin Norfloxacino Floxacin Norfloxacino Floxacin Norfloxacino Floxacin Norfloxacino Floxacin Norfloxacino Floxacin Norfloxacino Floxacin
186
ANEXO C PARECER PARA AMOXICILINA DO COMIT DE TICA EM PESQUISA
187
ANEXO D PARECER PARA NORFLOXACINO DO COMIT DE TICA EM PESQUISA
188
ANEXO E PARECER PARA OXCARBAZEPINA DO COMIT DE TICA EM PESQUISA
189
ANEXO F CERTIFICADO DE QUALIDADE DO PADRO ANALTICO AMOXICILINA
190
ANEXO G CERTIFICADO DE QUALIDADE DO PADRO ANALTICO CEFADROXIL
191
ANEXO H CERTIFICADO DE QUALIDADE DO PADRO ANALTICO NORFLOXACINO
192
ANEXO I CERTIFICADO DE QUALIDADE DO PADRO ANALTICO CIPROFLOXACINO
193
ANEXO J CERTIFICADO DE QUALIDADE DO PADRO ANALTICO OXCARBAZEPINA
194
ANEXO L CERTIFICADO DE QUALIDADE DO PADRO ANALTICO MHD
195
ANEXO M CERTIFICADO DE QUALIDADE DO PADRO ANALTICO CARBAMAZEPINA
Das könnte Ihnen auch gefallen
- Constipacao Intestinal 2011Dokument29 SeitenConstipacao Intestinal 2011Ismael LeiteNoch keine Bewertungen
- Controle RespiratorioDokument21 SeitenControle RespiratorioIsmael LeiteNoch keine Bewertungen
- Constipaçao e DiarreiaDokument13 SeitenConstipaçao e DiarreiaIsmael LeiteNoch keine Bewertungen
- Fisiologia Cardiaca FARM2010Dokument25 SeitenFisiologia Cardiaca FARM2010Ismael LeiteNoch keine Bewertungen
- Aula 02 Principios Da BioeticaDokument27 SeitenAula 02 Principios Da BioeticaIsmael LeiteNoch keine Bewertungen
- Bioeletrogênse 2010Dokument28 SeitenBioeletrogênse 2010Ismael LeiteNoch keine Bewertungen
- AminoglicosídeosDokument12 SeitenAminoglicosídeosIsmael LeiteNoch keine Bewertungen
- Agentes Inotrópicos Positivos.Dokument47 SeitenAgentes Inotrópicos Positivos.Ismael Leite100% (1)
- Dissertação Ismael PDFDokument197 SeitenDissertação Ismael PDFIsmael LeiteNoch keine Bewertungen
- Dissertação Ismael PDFDokument197 SeitenDissertação Ismael PDFIsmael LeiteNoch keine Bewertungen
- Agentes Inotrópicos Positivos.Dokument47 SeitenAgentes Inotrópicos Positivos.Ismael Leite100% (1)
- NirDokument7 SeitenNirDaniella DenleschiNoch keine Bewertungen
- Aula de Cromatografia Gasosa e GC/MSDokument88 SeitenAula de Cromatografia Gasosa e GC/MSGabriel SoaresNoch keine Bewertungen
- 2022 03 15 18 33 29 65407500 Eletromagnetismo Parte IDokument90 Seiten2022 03 15 18 33 29 65407500 Eletromagnetismo Parte IBruno SilvaNoch keine Bewertungen
- Lista 16Dokument15 SeitenLista 16Lucas HenriqueNoch keine Bewertungen
- Espectrometria de Massas 2Dokument14 SeitenEspectrometria de Massas 2Mariane Gabriela Cesar Ribeiro FerreiraNoch keine Bewertungen
- ICP Techiques Detailed MIA MCQDokument99 SeitenICP Techiques Detailed MIA MCQTiago PachecoNoch keine Bewertungen
- 2 - Aprofundado Magnetismo - Força MagnéticaDokument16 Seiten2 - Aprofundado Magnetismo - Força MagnéticaRaquel HernándezNoch keine Bewertungen
- Pólen de Abelha Como Bioindicador de Contaminação Ambiental Por Pesticidas - ScienceDirectDokument18 SeitenPólen de Abelha Como Bioindicador de Contaminação Ambiental Por Pesticidas - ScienceDirectTony PereiraNoch keine Bewertungen
- Teste de Estanqueidade - Mecatrônica Atual - Automação Industrial de Processos e ManufaturaDokument8 SeitenTeste de Estanqueidade - Mecatrônica Atual - Automação Industrial de Processos e ManufaturaLecoXavierNoch keine Bewertungen
- Emissão Atômica 2023Dokument60 SeitenEmissão Atômica 2023Kauan TelesNoch keine Bewertungen
- Apostila Espectrometria de Massas - Débora AzevedoDokument106 SeitenApostila Espectrometria de Massas - Débora AzevedoVinícius Benedetti100% (1)
- CromatografiaDokument103 SeitenCromatografiaValterF.JuscelinoNoch keine Bewertungen
- Espectrometria de MassaDokument2 SeitenEspectrometria de MassaIMVG DatabaseNoch keine Bewertungen
- Espectrometria de MassasDokument11 SeitenEspectrometria de MassasJoão Wilson FilhoNoch keine Bewertungen
- Cópia de Trab - MetodologiaDokument4 SeitenCópia de Trab - MetodologiaLorraine Alves da SilvaNoch keine Bewertungen
- Ebook Oil Essentials Origins WorkingDokument24 SeitenEbook Oil Essentials Origins WorkingVENDAS SULVEDANoch keine Bewertungen
- (CEFARMA) MASSAS - Junho 2012 - IMPRESSAO PDFDokument35 Seiten(CEFARMA) MASSAS - Junho 2012 - IMPRESSAO PDFGrace FernandesNoch keine Bewertungen
- Diluição Isotópica - EltonDokument33 SeitenDiluição Isotópica - Eltonapi-3761906100% (1)
- Identificação de THC em Semente de MaconhaDokument49 SeitenIdentificação de THC em Semente de MaconhaCadu OliveiraNoch keine Bewertungen
- GRAVIMETRIA OU ANLISE GRAVIMTRICA - Parte 1Dokument24 SeitenGRAVIMETRIA OU ANLISE GRAVIMTRICA - Parte 1pedro bulhoes100% (1)
- Abnt 13407 1995Dokument8 SeitenAbnt 13407 1995Fernando FakwNoch keine Bewertungen
- Incêndios de Petróleo e PetroquimicoDokument267 SeitenIncêndios de Petróleo e PetroquimicogiracamposNoch keine Bewertungen
- CG EM Cromatografia Gasosa Acoplada A Espectrometro de Massas PDFDokument62 SeitenCG EM Cromatografia Gasosa Acoplada A Espectrometro de Massas PDFmateus_laguardiaNoch keine Bewertungen
- CPTG - Certificado de Pureza e Grau Terapêutico - A Diferença DōTERRADokument2 SeitenCPTG - Certificado de Pureza e Grau Terapêutico - A Diferença DōTERRAAmor da Terra100% (1)
- Resolução Do TópicoooosDokument17 SeitenResolução Do TópicoooosVânia Nazaré Maia Dos Santos86% (7)
- Bachelard - o Filósofo Da DesilusãoDokument26 SeitenBachelard - o Filósofo Da DesilusãoMauricio MeloNoch keine Bewertungen
- FragmentaçãoDokument107 SeitenFragmentaçãojudaviesNoch keine Bewertungen
- E Book Análise de Falhas em Peças PoliméricasDokument68 SeitenE Book Análise de Falhas em Peças PoliméricasÉrica Rodrigues100% (1)
- O Estado Da Arte Da Cromatografia Associada À Espectrometria de Massas Acoplada ÀDokument14 SeitenO Estado Da Arte Da Cromatografia Associada À Espectrometria de Massas Acoplada Àguilherme.patricioNoch keine Bewertungen
- Aulas Massa para AlunosDokument27 SeitenAulas Massa para AlunosCarol MantovaniNoch keine Bewertungen