Das könnte Ihnen auch gefallen
- Special and Abbreviated 510 (K)Dokument18 SeitenSpecial and Abbreviated 510 (K)hemkenbpNoch keine Bewertungen
- NEWS CENTER Maine (NCM) Sent A List of Questions To The FDA and These Were Their ResponsesDokument2 SeitenNEWS CENTER Maine (NCM) Sent A List of Questions To The FDA and These Were Their ResponsesNEWS CENTER MaineNoch keine Bewertungen
- Classify Your Medical DeviceDokument3 SeitenClassify Your Medical DeviceChris HartoyoNoch keine Bewertungen
- Medical Devices and IVDs: Fit for the new EU-Regulations: Your complete seminar for projekt, study and jobVon EverandMedical Devices and IVDs: Fit for the new EU-Regulations: Your complete seminar for projekt, study and jobNoch keine Bewertungen
- Data Integrity and Compliance: A Primer for Medical Product ManufacturersVon EverandData Integrity and Compliance: A Primer for Medical Product ManufacturersNoch keine Bewertungen
- US Prep 510k Submission White Paper EMERGODokument10 SeitenUS Prep 510k Submission White Paper EMERGORamboNoch keine Bewertungen
- Biometrix - FDA-510K and Usability StudiesDokument17 SeitenBiometrix - FDA-510K and Usability StudiesgabababaNoch keine Bewertungen
- Biocompatibility Protocols for Medical Devices and MaterialsVon EverandBiocompatibility Protocols for Medical Devices and MaterialsNoch keine Bewertungen
- European Union Regulation of in Vitro Diagnostic Medical DevicesDokument26 SeitenEuropean Union Regulation of in Vitro Diagnostic Medical DevicesLuis Arístides Torres SánchezNoch keine Bewertungen
- Medical Device Reporting A Complete Guide - 2020 EditionVon EverandMedical Device Reporting A Complete Guide - 2020 EditionNoch keine Bewertungen
- Scientific Advice Meetings: A Guide to Successful Interactions with FDA, EMA and BeyondVon EverandScientific Advice Meetings: A Guide to Successful Interactions with FDA, EMA and BeyondNoch keine Bewertungen
- Careers in Focus: Pharmaceuticals and Biotechnology, Third EditionVon EverandCareers in Focus: Pharmaceuticals and Biotechnology, Third EditionNoch keine Bewertungen
- Medical Device Reporting System A Complete Guide - 2020 EditionVon EverandMedical Device Reporting System A Complete Guide - 2020 EditionNoch keine Bewertungen
- Leachables and Extractables Handbook: Safety Evaluation, Qualification, and Best Practices Applied to Inhalation Drug ProductsVon EverandLeachables and Extractables Handbook: Safety Evaluation, Qualification, and Best Practices Applied to Inhalation Drug ProductsDouglas J. BallNoch keine Bewertungen
- The Survival Guide to EU Medical Device RegulationsVon EverandThe Survival Guide to EU Medical Device RegulationsBewertung: 5 von 5 Sternen5/5 (1)
- FDA Guidance - 510 K ChecklistDokument3 SeitenFDA Guidance - 510 K ChecklistHila Cohen100% (3)
- Clinical Evaluation As Per CE MarkingDokument4 SeitenClinical Evaluation As Per CE MarkingSaraNoch keine Bewertungen
- Medical Device Software Strategy A Complete Guide - 2020 EditionVon EverandMedical Device Software Strategy A Complete Guide - 2020 EditionBewertung: 5 von 5 Sternen5/5 (1)
- ISO 14971 A Complete Guide - 2020 EditionVon EverandISO 14971 A Complete Guide - 2020 EditionBewertung: 2 von 5 Sternen2/5 (1)
- ISO 14971 A Complete Guide - 2021 EditionVon EverandISO 14971 A Complete Guide - 2021 EditionBewertung: 1 von 5 Sternen1/5 (1)
- Statistical Methods for Evaluating Safety in Medical Product DevelopmentVon EverandStatistical Methods for Evaluating Safety in Medical Product DevelopmentA. Lawrence GouldNoch keine Bewertungen
- 8 Key Changes To Understand in The New European MDR and IVDRDokument6 Seiten8 Key Changes To Understand in The New European MDR and IVDRKabomed QANoch keine Bewertungen
- Portfolio, Program, and Project Management in the Pharmaceutical and Biotechnology IndustriesVon EverandPortfolio, Program, and Project Management in the Pharmaceutical and Biotechnology IndustriesPete HarpumNoch keine Bewertungen
- Managing to the New Regulatory Reality: Doing Business Under the Dodd-Frank ActVon EverandManaging to the New Regulatory Reality: Doing Business Under the Dodd-Frank ActNoch keine Bewertungen
- Good Distribution Practices A Complete Guide - 2021 EditionVon EverandGood Distribution Practices A Complete Guide - 2021 EditionNoch keine Bewertungen
- Leitfaden Fuer App Entwickler enDokument65 SeitenLeitfaden Fuer App Entwickler enloipoilNoch keine Bewertungen
- ISO 13485 Quality Management System A Complete Guide - 2020 EditionVon EverandISO 13485 Quality Management System A Complete Guide - 2020 EditionNoch keine Bewertungen
- 510 (K) Format Guidance, Including Standards Form, and Extensions Clinical Trial Form and 510 (K) PDFDokument27 Seiten510 (K) Format Guidance, Including Standards Form, and Extensions Clinical Trial Form and 510 (K) PDFMichael wangNoch keine Bewertungen
- Medical Device Connectivity The Ultimate Step-By-Step GuideVon EverandMedical Device Connectivity The Ultimate Step-By-Step GuideNoch keine Bewertungen
- Abbreviated 510k - When The Abbreviation Is AllowedDokument5 SeitenAbbreviated 510k - When The Abbreviation Is AllowedRegulatonomous OpenNoch keine Bewertungen
- Australia Post Market Activity GuidelinesDokument31 SeitenAustralia Post Market Activity Guidelinesspenceblack7999Noch keine Bewertungen
- Brazil GMP Compliance White PaperDokument11 SeitenBrazil GMP Compliance White PaperTomasz WojteraNoch keine Bewertungen
- Bsi Smart Support Post Market SurveillanceDokument10 SeitenBsi Smart Support Post Market SurveillanceMauro CostaNoch keine Bewertungen
- Usability Engineering ReportDokument2 SeitenUsability Engineering ReportdocuNoch keine Bewertungen
- Pharmacovigilance Medical Writing: A Good Practice GuideVon EverandPharmacovigilance Medical Writing: A Good Practice GuideBewertung: 4 von 5 Sternen4/5 (1)
- Db10g Rac Exam Study Guide 320804Dokument5 SeitenDb10g Rac Exam Study Guide 320804Anand ShankarNoch keine Bewertungen
- Medical Device Reporting A Complete Guide - 2019 EditionVon EverandMedical Device Reporting A Complete Guide - 2019 EditionNoch keine Bewertungen
- Medical Device Software Quality Management A Complete Guide - 2020 EditionVon EverandMedical Device Software Quality Management A Complete Guide - 2020 EditionNoch keine Bewertungen
- Clinical Evaluation and Investigation of Medical Devices under the new EU-RegulationVon EverandClinical Evaluation and Investigation of Medical Devices under the new EU-RegulationBewertung: 5 von 5 Sternen5/5 (1)
- An Overview of FDA Regulated Products: From Drugs and Cosmetics to Food and TobaccoVon EverandAn Overview of FDA Regulated Products: From Drugs and Cosmetics to Food and TobaccoEunjoo PacificiBewertung: 5 von 5 Sternen5/5 (1)
- Advances in Cancer Nanotheranostics for Experimental and Personalized MedicineVon EverandAdvances in Cancer Nanotheranostics for Experimental and Personalized MedicineNoch keine Bewertungen
- Risk Assessment Report - Proposal & Annotated BibliographyDokument7 SeitenRisk Assessment Report - Proposal & Annotated BibliographyDaniel KounahNoch keine Bewertungen
- Protocol Template 05feb2016 508Dokument3 SeitenProtocol Template 05feb2016 508Dwi Annisa AmaliaSariNoch keine Bewertungen
- Bsi MD MDR Readiness Review Es enDokument9 SeitenBsi MD MDR Readiness Review Es enIAS IndiaNoch keine Bewertungen
- Cybersecurity of Medical DevicesDokument12 SeitenCybersecurity of Medical DevicesppisupaNoch keine Bewertungen
- Regulatory Aspects of Pharmaceutical Quality System: Brief IntroductionVon EverandRegulatory Aspects of Pharmaceutical Quality System: Brief IntroductionNoch keine Bewertungen
- Police Unions Lose Bid To Keep Disciplinary Records A SecretDokument3 SeitenPolice Unions Lose Bid To Keep Disciplinary Records A SecretJames LindonNoch keine Bewertungen
- Overture 28 Westminster Homosexuality ADsDokument2 SeitenOverture 28 Westminster Homosexuality ADsJames LindonNoch keine Bewertungen
- 09.06.2018 StarmanDokument7 Seiten09.06.2018 StarmanJames LindonNoch keine Bewertungen
- Overture 4 Calvary Nashville StatementDokument3 SeitenOverture 4 Calvary Nashville StatementJames LindonNoch keine Bewertungen
- 09.06.2018 WarmothDokument8 Seiten09.06.2018 WarmothJames LindonNoch keine Bewertungen
- Anderson ComplaintDokument15 SeitenAnderson ComplaintBasseemNoch keine Bewertungen
- Donald J. Trump IndictmentDokument16 SeitenDonald J. Trump IndictmentStefan Becket93% (27)
- State Of: Steven W. Schierholt, Esq. Executive Director John R. GaDokument6 SeitenState Of: Steven W. Schierholt, Esq. Executive Director John R. GaJames LindonNoch keine Bewertungen
- 10.04.2018 HartleyDokument8 Seiten10.04.2018 HartleyJames LindonNoch keine Bewertungen
- 10.10.2018 GomezDokument5 Seiten10.10.2018 GomezJames LindonNoch keine Bewertungen
- 09.06.2018 MiddlebrooksDokument5 Seiten09.06.2018 MiddlebrooksJames LindonNoch keine Bewertungen
- 10.09.2018 Medi-Mart PharmacyDokument6 Seiten10.09.2018 Medi-Mart PharmacyJames LindonNoch keine Bewertungen
- 09.14.2018 NgohDokument9 Seiten09.14.2018 NgohJames LindonNoch keine Bewertungen
- 10.10.2018 GrigsbyDokument5 Seiten10.10.2018 GrigsbyJames LindonNoch keine Bewertungen
- 8.6.18 Lia HarbDokument3 Seiten8.6.18 Lia HarbJames LindonNoch keine Bewertungen
- State Of: PendingDokument7 SeitenState Of: PendingJames LindonNoch keine Bewertungen
- 09.06.2018 Pamela ApplegateDokument6 Seiten09.06.2018 Pamela ApplegateJames LindonNoch keine Bewertungen
- 09.26.2018 LottDokument6 Seiten09.26.2018 LottJames LindonNoch keine Bewertungen
- Robert Garrity Prison RecordDokument1 SeiteRobert Garrity Prison RecordJames LindonNoch keine Bewertungen
- Sc-,:iegi?5: in The Matter Of: CASE NO. 2016-1909 License No. 02-1339450Dokument6 SeitenSc-,:iegi?5: in The Matter Of: CASE NO. 2016-1909 License No. 02-1339450James LindonNoch keine Bewertungen
- 10.02.2018 MascioDokument6 Seiten10.02.2018 MascioJames LindonNoch keine Bewertungen
- James Lindon Attorney, Ph.D. 35104 Saddle Creek Avon, Ohio 44011-4907Dokument1 SeiteJames Lindon Attorney, Ph.D. 35104 Saddle Creek Avon, Ohio 44011-4907James LindonNoch keine Bewertungen
- 8.23.18 Better Living Clinic AkronDokument5 Seiten8.23.18 Better Living Clinic AkronJames LindonNoch keine Bewertungen
- Robert Garrity Ohio Board of Pharmacy 05-13-2002 RevocationDokument19 SeitenRobert Garrity Ohio Board of Pharmacy 05-13-2002 RevocationJames LindonNoch keine Bewertungen
- 1.3.18 Ohio Board of Pharmacy Notice of Opportunity For Hearing James LindonDokument8 Seiten1.3.18 Ohio Board of Pharmacy Notice of Opportunity For Hearing James LindonJames LindonNoch keine Bewertungen
- 1.22.18 Ohio Board of Pharmacy Notice of Opportunity For Hearing James LindonDokument3 Seiten1.22.18 Ohio Board of Pharmacy Notice of Opportunity For Hearing James LindonJames LindonNoch keine Bewertungen
- Robert J Garrity Felony Conviction Cr-01-406023-ZaDokument2 SeitenRobert J Garrity Felony Conviction Cr-01-406023-ZaJames LindonNoch keine Bewertungen
- Robert J Garrity Felony Conviction Cr-01-406023-ZaDokument2 SeitenRobert J Garrity Felony Conviction Cr-01-406023-ZaJames LindonNoch keine Bewertungen
- 1.25.18 Ohio Board of Pharmacy Notice of Opportunity For Hearing James LindonDokument13 Seiten1.25.18 Ohio Board of Pharmacy Notice of Opportunity For Hearing James LindonJames LindonNoch keine Bewertungen
- Jose Garcia Verdict 11-18-2015 James Lindon PDFDokument3 SeitenJose Garcia Verdict 11-18-2015 James Lindon PDFJames LindonNoch keine Bewertungen
- Jntu Hyderabad - 2010 Dec - 07a51404 Kinematicsofmachineryfr 3180Dokument10 SeitenJntu Hyderabad - 2010 Dec - 07a51404 Kinematicsofmachineryfr 3180bbkNoch keine Bewertungen
- IPv6 RoutingDokument27 SeitenIPv6 RoutingTtmManNoch keine Bewertungen
- PDFDokument2 SeitenPDFSalim AshorNoch keine Bewertungen
- Malhotra MR6e 02Dokument29 SeitenMalhotra MR6e 02Mohsin Ali RazaNoch keine Bewertungen
- Thought Leadership Is The New Sales PitchDokument8 SeitenThought Leadership Is The New Sales PitchChad NelsonNoch keine Bewertungen
- HIU Range Design GuideDokument24 SeitenHIU Range Design Guidesachinsaklani23Noch keine Bewertungen
- Angoca Db2 Cheat Sheet For DevelopmentDokument3 SeitenAngoca Db2 Cheat Sheet For DevelopmentTanveer AhmedNoch keine Bewertungen
- PCBA-WP543HV HW Manual Rev1.05Dokument21 SeitenPCBA-WP543HV HW Manual Rev1.05Surapong Pongchaiprateep100% (1)
- Mobile Networks and ApplicationsDokument232 SeitenMobile Networks and ApplicationsrabrajNoch keine Bewertungen
- Kinematics of MachinesDokument2 SeitenKinematics of MachinesBhavesh J MakwanaNoch keine Bewertungen
- Dap 018 ADokument28 SeitenDap 018 AajoaomvNoch keine Bewertungen
- What Is A Bioclimatic Chart - EHowDokument2 SeitenWhat Is A Bioclimatic Chart - EHowonkhgfg kjhh jghNoch keine Bewertungen
- Failure Mechanisms of C-Steels (API 571)Dokument90 SeitenFailure Mechanisms of C-Steels (API 571)Abdul Gafoor Shaikh50% (2)
- Truss Operating Manual: Version 7aDokument28 SeitenTruss Operating Manual: Version 7adoyoudeNoch keine Bewertungen
- Dimetra Ip CompactDokument2 SeitenDimetra Ip CompactGrompolLopmorgNoch keine Bewertungen
- Tema Tubesheet Calculation SheetDokument1 SeiteTema Tubesheet Calculation SheetSanjeev KachharaNoch keine Bewertungen
- Usermanual en Manual Arium Bagtanks WH26010 ADokument33 SeitenUsermanual en Manual Arium Bagtanks WH26010 AԳոռ ԽաչատրյանNoch keine Bewertungen
- 4 Unit 2 - Solid State Welding (SSW)Dokument33 Seiten4 Unit 2 - Solid State Welding (SSW)Aditya Kumar100% (2)
- 50 58 Eng Concrete TestingDokument92 Seiten50 58 Eng Concrete TestingJimmy LopezNoch keine Bewertungen
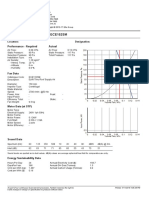
- Technical Data - Fan Model ECE152SM: Location: Designation: Performance - Required ActualDokument2 SeitenTechnical Data - Fan Model ECE152SM: Location: Designation: Performance - Required ActualJNoch keine Bewertungen
- CM Dashboard Arunachal PradeshDokument8 SeitenCM Dashboard Arunachal PradeshDebashish Goswami100% (1)
- Dell's Marketing Strategy - 2006Dokument58 SeitenDell's Marketing Strategy - 2006Preeti IyerNoch keine Bewertungen
- On Phase ChangesDokument28 SeitenOn Phase Changesapi-313517608Noch keine Bewertungen
- Tquins Resources Training Template For MT Work Programme SCS RevisedDokument12 SeitenTquins Resources Training Template For MT Work Programme SCS RevisedLileth LagasimNoch keine Bewertungen
- Prince CPVC Pipes and Fittings Pricelist1Dokument1 SeitePrince CPVC Pipes and Fittings Pricelist1Dee Raja38% (8)
- Performance of Trinidad Gas Reservoirs PDFDokument11 SeitenPerformance of Trinidad Gas Reservoirs PDFMarcus ChanNoch keine Bewertungen
- Accomplishment Report in ESPDokument7 SeitenAccomplishment Report in ESPAldrin Perez85% (39)
- 4 Litre Closed SamplersDokument3 Seiten4 Litre Closed Samplerslimhockkin3766Noch keine Bewertungen
- Electoral ListDokument189 SeitenElectoral ListAhmadShazebAzharNoch keine Bewertungen
- StarbucksDokument19 SeitenStarbucksNimah SaeedNoch keine Bewertungen