Das könnte Ihnen auch gefallen
- Kinetics and Related Engineering Aspects of Catalytic Liquid-Phase Oxidation of P-Xylene To Terephthalic AcidDokument17 SeitenKinetics and Related Engineering Aspects of Catalytic Liquid-Phase Oxidation of P-Xylene To Terephthalic AcidAbdullah JavedNoch keine Bewertungen
- Water Gas Shift Reaction Kinetics and Reactor Modeling For Fuel Cell Grade Hydrogen PDFDokument8 SeitenWater Gas Shift Reaction Kinetics and Reactor Modeling For Fuel Cell Grade Hydrogen PDFKmilo BolañosNoch keine Bewertungen
- Gar CA Molina 2007Dokument7 SeitenGar CA Molina 2007LuciaMarinaR.OrizaNoch keine Bewertungen
- 1 s2.0 S036054421500910X AmDokument32 Seiten1 s2.0 S036054421500910X AmnahomNoch keine Bewertungen
- Example PDFDokument14 SeitenExample PDFJuan PerezNoch keine Bewertungen
- Experimental and Theoretical Investigation GAS PUTITYDokument167 SeitenExperimental and Theoretical Investigation GAS PUTITYJesús Javier Antuña CouceiroNoch keine Bewertungen
- Behin Et Al-2013-Chemical Engineering & TechnologyDokument10 SeitenBehin Et Al-2013-Chemical Engineering & TechnologyIgnacio JuanNoch keine Bewertungen
- A Comparative Study of Non-Oxidative Pyrolysis and Oxidative Cracking of Cyclohexane To Light AlkenesDokument17 SeitenA Comparative Study of Non-Oxidative Pyrolysis and Oxidative Cracking of Cyclohexane To Light AlkenesArif HidayatNoch keine Bewertungen
- 10.1007@s11144 020 01851 3Dokument17 Seiten10.1007@s11144 020 01851 3Badis GueloulNoch keine Bewertungen
- 2004 Methanol Steam Reforming Over CuZnOAl2O3 Catalyst Kinetics and Effectiveness FactorDokument11 Seiten2004 Methanol Steam Reforming Over CuZnOAl2O3 Catalyst Kinetics and Effectiveness FactorChauNoch keine Bewertungen
- Reaction Network and Kinetic Modeling of PDFDokument11 SeitenReaction Network and Kinetic Modeling of PDFGabriel BuftiaNoch keine Bewertungen
- Springer - 2001 - J. - Electrochem. - Soc. - 148 - A11Dokument14 SeitenSpringer - 2001 - J. - Electrochem. - Soc. - 148 - A11Faseeh KKNoch keine Bewertungen
- Proposition of A Minimum Set of Independent Chemical Reactions To Model Gas-Phase Composition During Gasification of Complex CokesDokument7 SeitenProposition of A Minimum Set of Independent Chemical Reactions To Model Gas-Phase Composition During Gasification of Complex CokesJavier FrancesconiNoch keine Bewertungen
- Sequential Simulation of Dense Oxygen Permeation Membrane Reactor For Hydrogen Production From Oxidative Steam Reforming of Ethanol With ASPEN PLUS 20Dokument8 SeitenSequential Simulation of Dense Oxygen Permeation Membrane Reactor For Hydrogen Production From Oxidative Steam Reforming of Ethanol With ASPEN PLUS 20serchNoch keine Bewertungen
- Plasma Hexane 2014 IEEE PDFDokument11 SeitenPlasma Hexane 2014 IEEE PDFCAMILA COBOS MOLANONoch keine Bewertungen
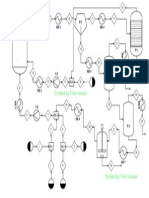
- A Conceptual Design of Novel Reactor For Liquid-Phase Oxidation of P-XyleneDokument2 SeitenA Conceptual Design of Novel Reactor For Liquid-Phase Oxidation of P-XyleneNoi Chem-EnNoch keine Bewertungen
- Numerical Simulation of An Industrial Fluid Catalytic Cracking RegeneratorDokument10 SeitenNumerical Simulation of An Industrial Fluid Catalytic Cracking RegeneratorsagarsrinivasNoch keine Bewertungen
- Aspen Plus Simulation of Saponification of Ethyl Acetate in The Presence of Sodium Hydroxide in A Plug Flow ReactorDokument8 SeitenAspen Plus Simulation of Saponification of Ethyl Acetate in The Presence of Sodium Hydroxide in A Plug Flow ReactorSoumajit SenNoch keine Bewertungen
- Project Descriptions 2012-13. Supplementary List 9.10.12Dokument7 SeitenProject Descriptions 2012-13. Supplementary List 9.10.12Moao AomoNoch keine Bewertungen
- Hydrogen Production Via Steam Reforming of Methane With Simultaneous Co Capture Over Cao - Ca Al ODokument7 SeitenHydrogen Production Via Steam Reforming of Methane With Simultaneous Co Capture Over Cao - Ca Al OMonica RoyNoch keine Bewertungen
- Reduced Mechanism Approach of Modeling Premixed Propane-Air Mixture Using ANSYS FluentDokument20 SeitenReduced Mechanism Approach of Modeling Premixed Propane-Air Mixture Using ANSYS FluentDinesh TPNoch keine Bewertungen
- GPC3 - CO2 Capture Using An Aqueous Formulated Solvent - Published - June 2015Dokument9 SeitenGPC3 - CO2 Capture Using An Aqueous Formulated Solvent - Published - June 2015Rahul BhosaleNoch keine Bewertungen
- Experimental and Modeling Study On Zeolite Catalysts For Diesel EnginesDokument10 SeitenExperimental and Modeling Study On Zeolite Catalysts For Diesel Enginesthai avvaiNoch keine Bewertungen
- Optimisation of Photo-Fenton-Like Degradation of Aqueous Polyacrylic Acid Using Box-Behnken Experimental DesignDokument12 SeitenOptimisation of Photo-Fenton-Like Degradation of Aqueous Polyacrylic Acid Using Box-Behnken Experimental DesignRaphael BrigagãoNoch keine Bewertungen
- Methane Steam ReformingDokument118 SeitenMethane Steam Reformingrezaroohollahi100% (2)
- Chemical Engineering Journal (A Kinetic and Process Modeling Study of CO2 Capture With MEA-Promoted)Dokument9 SeitenChemical Engineering Journal (A Kinetic and Process Modeling Study of CO2 Capture With MEA-Promoted)Febrian Adhitya RachmanNoch keine Bewertungen
- Hydrogen Production From Fossil and Biomass Fuels: Catalyst Development, Kinetics, and Reactor ModelingDokument4 SeitenHydrogen Production From Fossil and Biomass Fuels: Catalyst Development, Kinetics, and Reactor ModelingMazen OthmanNoch keine Bewertungen
- A Physical Absorption Process For The Capture of Co From Co - Rich Natural Gas StreamsDokument6 SeitenA Physical Absorption Process For The Capture of Co From Co - Rich Natural Gas StreamssinhleprovietNoch keine Bewertungen
- Journal of CO Utilization: Nora Meiri, Roman Radus, Moti HerskowitzDokument6 SeitenJournal of CO Utilization: Nora Meiri, Roman Radus, Moti HerskowitzFarah Talib Al-sudaniNoch keine Bewertungen
- Donazzi 2008Dokument18 SeitenDonazzi 2008Monika MartinezNoch keine Bewertungen
- Design and Control of A para Xylene by Air Oxidation ProcessDokument20 SeitenDesign and Control of A para Xylene by Air Oxidation ProcessSarat Na NiNoch keine Bewertungen
- Catalytic Transformations of CO2HDokument9 SeitenCatalytic Transformations of CO2Hbugaboo16Noch keine Bewertungen
- Plasma-Controlled Chemistry in Plasma Reforming of MethaneDokument10 SeitenPlasma-Controlled Chemistry in Plasma Reforming of MethaneViệtDũng TôNoch keine Bewertungen
- Modelling of Nutrient Removal Processes in An Intermittently Aerated BioreactorDokument8 SeitenModelling of Nutrient Removal Processes in An Intermittently Aerated BioreactorSalsa_Picante_BabyNoch keine Bewertungen
- Full 71094Dokument8 SeitenFull 71094Peter TaylorNoch keine Bewertungen
- Natural Gas As Feedstock For Fertilizer: A Thesis Submitted in Partial Fulfillment of The Requirements For The Degree ofDokument65 SeitenNatural Gas As Feedstock For Fertilizer: A Thesis Submitted in Partial Fulfillment of The Requirements For The Degree oframachandran_chemNoch keine Bewertungen
- 1 s2.0 S0009250910004860 MainDokument12 Seiten1 s2.0 S0009250910004860 MainFares EhabNoch keine Bewertungen
- Chenoweth 2008Dokument14 SeitenChenoweth 2008Omar HMNoch keine Bewertungen
- 6 Chilev 463-474Dokument12 Seiten6 Chilev 463-474emad hayekNoch keine Bewertungen
- Green Chemistry: Accepted ManuscriptDokument10 SeitenGreen Chemistry: Accepted ManuscriptSyarif HidayatNoch keine Bewertungen
- Naphtha Steam Reforming For Hydrogen ProductionDokument9 SeitenNaphtha Steam Reforming For Hydrogen ProductionsatishchemengNoch keine Bewertungen
- Design of Reactive Distillations For Acetic Acid EsterificationDokument17 SeitenDesign of Reactive Distillations For Acetic Acid Esterificationehsan zeraatkarNoch keine Bewertungen
- Absorption Tower For CO2Dokument11 SeitenAbsorption Tower For CO2ankitsamriaNoch keine Bewertungen
- Content ServerDokument10 SeitenContent ServerPaco CeronNoch keine Bewertungen
- Qua 25497Dokument12 SeitenQua 25497snehasis banerjeeNoch keine Bewertungen
- AbsorçãoDokument8 SeitenAbsorçãoSamanta De Jesus FerreiraNoch keine Bewertungen
- Artigo 1Dokument7 SeitenArtigo 1Rafael AmaranteNoch keine Bewertungen
- Automotive Exhaust Gas Conversion - From Elementary Step Kinetics To Prediction of Emission DynamicsDokument9 SeitenAutomotive Exhaust Gas Conversion - From Elementary Step Kinetics To Prediction of Emission DynamicsAndang Hastu PNoch keine Bewertungen
- Processes 07 00136Dokument22 SeitenProcesses 07 00136vanessa jimenezNoch keine Bewertungen
- PMR v19 I1 012 014Dokument3 SeitenPMR v19 I1 012 014Arianne Jayne G. GubaNoch keine Bewertungen
- Carbon Dioxide Capture For The Oxidative Coupling of Methane Process - A Case Study in Mini-Plant Scale - Repke-Stunkel Paper Stuenkel Repke Mini-PlantDokument10 SeitenCarbon Dioxide Capture For The Oxidative Coupling of Methane Process - A Case Study in Mini-Plant Scale - Repke-Stunkel Paper Stuenkel Repke Mini-PlantZheqi YuNoch keine Bewertungen
- A Technical and Economic Assessment of Ammonia-Based Post-Combustion CO2 Capture at Coal-Fired Power PlantsDokument10 SeitenA Technical and Economic Assessment of Ammonia-Based Post-Combustion CO2 Capture at Coal-Fired Power PlantsBánh Cuốn Tôm ThịtNoch keine Bewertungen
- 2008 Minh AICHE PDFDokument9 Seiten2008 Minh AICHE PDFinf2014Noch keine Bewertungen
- Modeling and Application of Response Surface Methodology in Optimization of A Commercial Continuous Catalytic Reforming ProcessDokument21 SeitenModeling and Application of Response Surface Methodology in Optimization of A Commercial Continuous Catalytic Reforming ProcessArash AbbasiNoch keine Bewertungen
- Gas-Phase Mass Transfer Coefficient of CO in Different Alkanolamine Solutions Within Packed-Bed Absorption ColumnDokument10 SeitenGas-Phase Mass Transfer Coefficient of CO in Different Alkanolamine Solutions Within Packed-Bed Absorption ColumnHamza AliNoch keine Bewertungen
- 10 1016@j Cep 2017 03 022Dokument24 Seiten10 1016@j Cep 2017 03 022Violeta GarciaNoch keine Bewertungen
- Reaction RatesDokument8 SeitenReaction RatesAbolhasan AmeriNoch keine Bewertungen
- Introduction to Supercritical Fluids: A Spreadsheet-based ApproachVon EverandIntroduction to Supercritical Fluids: A Spreadsheet-based ApproachNoch keine Bewertungen
- Carbon Capture Technologies for Gas-Turbine-Based Power PlantsVon EverandCarbon Capture Technologies for Gas-Turbine-Based Power PlantsNoch keine Bewertungen
- Direct Methane to Methanol: Foundations and Prospects of the ProcessVon EverandDirect Methane to Methanol: Foundations and Prospects of the ProcessNoch keine Bewertungen
- Nitrogen BlanketingDokument7 SeitenNitrogen Blanketing3bandhuNoch keine Bewertungen
- Parker Tank Blanketing White Paper PDFDokument4 SeitenParker Tank Blanketing White Paper PDFshashi kant kumarNoch keine Bewertungen
- Dow Filmtec BW30XFR 400 34iDokument2 SeitenDow Filmtec BW30XFR 400 34iJuan Camilo HenaoNoch keine Bewertungen
- Uniteolstates Patent Office: "133.2535.lftii?iiii'tfiiiw 7 'Dokument2 SeitenUniteolstates Patent Office: "133.2535.lftii?iiii'tfiiiw 7 'Juan Camilo HenaoNoch keine Bewertungen
- Batista 2013Dokument13 SeitenBatista 2013Juan Camilo HenaoNoch keine Bewertungen
- Purge With NitrogenDokument5 SeitenPurge With Nitrogendeion29100% (1)
- When You Were A Child, What Job Did You Want To Do When You Grew Up?Dokument1 SeiteWhen You Were A Child, What Job Did You Want To Do When You Grew Up?Juan Camilo HenaoNoch keine Bewertungen
- Dow Filmtec BW30XFR 400 34iDokument2 SeitenDow Filmtec BW30XFR 400 34iJuan Camilo HenaoNoch keine Bewertungen
- Papper Co2Dokument15 SeitenPapper Co2Juan Camilo HenaoNoch keine Bewertungen
- Creative Science & Research - Screen Printing Press (2004)Dokument21 SeitenCreative Science & Research - Screen Printing Press (2004)amrandconan100% (2)
- Algebra Angel Cap9Dokument66 SeitenAlgebra Angel Cap9Ariel GomezNoch keine Bewertungen
- Learn English Podcasts Elementary 01 01 Support Pack TranscriptDokument22 SeitenLearn English Podcasts Elementary 01 01 Support Pack Transcriptpramithbuddika100% (1)
- Created by Trial Version: HE-3 HE-4Dokument1 SeiteCreated by Trial Version: HE-3 HE-4Juan Camilo HenaoNoch keine Bewertungen
- Zirconia-Supported Vanadium Oxide Catalysts For Ammoxidation and Oxidation of Toluene: A Characterization and Activity StudyDokument22 SeitenZirconia-Supported Vanadium Oxide Catalysts For Ammoxidation and Oxidation of Toluene: A Characterization and Activity StudyJuan Camilo HenaoNoch keine Bewertungen
- Extreme Independence - All Combinations of 4 LimbsDokument3 SeitenExtreme Independence - All Combinations of 4 LimbsJuan Camilo HenaoNoch keine Bewertungen
- IT H Unit IGDokument19 SeitenIT H Unit IGJuan Camilo HenaoNoch keine Bewertungen
- CLASS 300 Welding Neck & Slip-On Flanges: ASTM A-105 & A181-CLASS 60Dokument3 SeitenCLASS 300 Welding Neck & Slip-On Flanges: ASTM A-105 & A181-CLASS 60Juan Camilo HenaoNoch keine Bewertungen
- Benzoguanamine: Product Specification SheetDokument1 SeiteBenzoguanamine: Product Specification SheetJuan Camilo HenaoNoch keine Bewertungen
- 1 PBDokument11 Seiten1 PBJuan Camilo HenaoNoch keine Bewertungen
- 02bfe50dacb1fe39e0000000 PDFDokument50 Seiten02bfe50dacb1fe39e0000000 PDFJuan Camilo HenaoNoch keine Bewertungen
- Ie00064a001 PDFDokument9 SeitenIe00064a001 PDFJuan Camilo HenaoNoch keine Bewertungen
- Aspen HYSYS DynamicsDokument3 SeitenAspen HYSYS DynamicsJuan Camilo HenaoNoch keine Bewertungen
- United States' Patent Office: Patented July 20, 1948Dokument5 SeitenUnited States' Patent Office: Patented July 20, 1948Juan Camilo HenaoNoch keine Bewertungen
- Flash ConceptsDokument2 SeitenFlash ConceptsJuan Camilo HenaoNoch keine Bewertungen
- Benzoguanamine: Product Specification SheetDokument1 SeiteBenzoguanamine: Product Specification SheetJuan Camilo HenaoNoch keine Bewertungen
- Poços de CaldasDokument8 SeitenPoços de CaldasAnninha FlavioNoch keine Bewertungen
- (Doi 10.1021/ie50289a025) M. Souders G. G. Brown - Design of Fractionating Columns I. Entrainment and Capacity PDFDokument6 Seiten(Doi 10.1021/ie50289a025) M. Souders G. G. Brown - Design of Fractionating Columns I. Entrainment and Capacity PDFJuan Camilo HenaoNoch keine Bewertungen
- Dowtherm A PropertiesDokument2 SeitenDowtherm A Propertiesflashiitm100% (1)
- AbsorptionStripping PDFDokument25 SeitenAbsorptionStripping PDFJuan Camilo HenaoNoch keine Bewertungen
- Fertilizers and Chemicals Travancore LTD: FACT ForwardDokument35 SeitenFertilizers and Chemicals Travancore LTD: FACT ForwardfharooksNoch keine Bewertungen
- Selective Hydrogenation of Phenol: Chemnanomat February 2018Dokument21 SeitenSelective Hydrogenation of Phenol: Chemnanomat February 2018Deo TolloNoch keine Bewertungen
- FYP ProposalDokument11 SeitenFYP ProposalArslan SamNoch keine Bewertungen
- Government Engineering College Bhuj: Gujarat Technological UniversityDokument23 SeitenGovernment Engineering College Bhuj: Gujarat Technological UniversityPâtël PrãtïkNoch keine Bewertungen
- Adipic AcidDokument33 SeitenAdipic AcidjY-renNoch keine Bewertungen
- A01 269Dokument11 SeitenA01 269icingrockNoch keine Bewertungen
- Sulfonation of Aniline ExperimentDokument3 SeitenSulfonation of Aniline Experimentpakpolitics206Noch keine Bewertungen
- Process-Design Intensification - Direct Synthesis of Adipic Acid in FlowDokument7 SeitenProcess-Design Intensification - Direct Synthesis of Adipic Acid in FlowMary Grace VelitarioNoch keine Bewertungen
- Apendice 1Dokument14 SeitenApendice 1JA UMAN1Noch keine Bewertungen
- Ullmann S Enc of Industrial Chemistry PLANTA PDFDokument12 SeitenUllmann S Enc of Industrial Chemistry PLANTA PDFTaylor PennaNoch keine Bewertungen
- CaprolactamDokument4 SeitenCaprolactamArchie HisolerNoch keine Bewertungen
- Dielectric Constants of MaterialsDokument68 SeitenDielectric Constants of MaterialsRUBEN DARIO BUCHELLYNoch keine Bewertungen
- Base Impacts® Data Documentation Sector: Plastics, Resins, Fibers and Fillers ProductionDokument44 SeitenBase Impacts® Data Documentation Sector: Plastics, Resins, Fibers and Fillers ProductionGerogesNoch keine Bewertungen
- Experiment No. 7 Oxidation of A Secondary Alcohol: Cyclohexanone From Cyclohexanol IDokument10 SeitenExperiment No. 7 Oxidation of A Secondary Alcohol: Cyclohexanone From Cyclohexanol IChaa UbaldoNoch keine Bewertungen
- BPP CO Product Costing v.0Dokument35 SeitenBPP CO Product Costing v.0fharooksNoch keine Bewertungen
- Ficha Cyclohexanone BASFDokument2 SeitenFicha Cyclohexanone BASFRicardo Javier PlasenciaNoch keine Bewertungen
- Oxidation of Cyclohexane and Ethylbenzene by Hydrogen Peroxide Over Co-Substituted Heteropolytungstate CatalystDokument6 SeitenOxidation of Cyclohexane and Ethylbenzene by Hydrogen Peroxide Over Co-Substituted Heteropolytungstate Catalystrungrawin ngamkhumNoch keine Bewertungen
- Caprolactam Process DescriptionDokument8 SeitenCaprolactam Process DescriptionJohannah Jane Abuel100% (2)
- Dielectric Constants of Common MaterialsDokument52 SeitenDielectric Constants of Common Materialsskattejag100% (1)
- Chen 1994Dokument6 SeitenChen 1994ZhalaNoch keine Bewertungen
- Cyclohexanol and CyclohexanoneDokument12 SeitenCyclohexanol and Cyclohexanonetatiana alvarezNoch keine Bewertungen
- Ullmann's Enc. of Industrial Chemistry PLANTA.Dokument12 SeitenUllmann's Enc. of Industrial Chemistry PLANTA.yoelarismendi100% (1)
- Preparation of Adipic Acid by Oxidation of Cyclohexanol and Cyclohexanone With Nitric Acid - Part 1. Reaction MechanismDokument17 SeitenPreparation of Adipic Acid by Oxidation of Cyclohexanol and Cyclohexanone With Nitric Acid - Part 1. Reaction MechanismEugenio Alejandro Pérez Reséndiz50% (2)
- Patente StamicarbonDokument6 SeitenPatente StamicarbonMiguelNoch keine Bewertungen
- Pan2004 Article One-StepSynthesisOfCyclohexanoDokument3 SeitenPan2004 Article One-StepSynthesisOfCyclohexanorenata guerreiroNoch keine Bewertungen
- Kapasitas Aseton Prasol ChemDokument59 SeitenKapasitas Aseton Prasol ChemMadeline Geovany TangaNoch keine Bewertungen
- (Topics in Chemical Engineering) Martyn S. Ray, David W. Johnston - Chemical Engineering Design Project - A Case Study Approach Topics in Chemical Engineering - Volume 6-Routledge (1989)Dokument2 Seiten(Topics in Chemical Engineering) Martyn S. Ray, David W. Johnston - Chemical Engineering Design Project - A Case Study Approach Topics in Chemical Engineering - Volume 6-Routledge (1989)Pratiwi Puji Ajeng LestariNoch keine Bewertungen
- Handbook of Thermodynamic Diagrams VOLUME2 PDFDokument409 SeitenHandbook of Thermodynamic Diagrams VOLUME2 PDFcarlitaGJNoch keine Bewertungen
- Production of Cyclohexanone From Phenol HydrogenationDokument157 SeitenProduction of Cyclohexanone From Phenol Hydrogenationnoman100% (2)
- Selectivity in Phenol Hydrogenation To Cyclohexanone Over Rh/Sio CatalystsDokument2 SeitenSelectivity in Phenol Hydrogenation To Cyclohexanone Over Rh/Sio CatalystsArun RNoch keine Bewertungen