Das könnte Ihnen auch gefallen
- Case Report Cushing SyndromeDokument8 SeitenCase Report Cushing SyndromeVivi DeviyanaNoch keine Bewertungen
- Adrenal IncidantelomaDokument22 SeitenAdrenal IncidantelomaAbdallah DarrasNoch keine Bewertungen
- Best Practice & Research Clinical Endocrinology & MetabolismDokument3 SeitenBest Practice & Research Clinical Endocrinology & Metabolismnyoman wianiNoch keine Bewertungen
- Cushing IncidDokument10 SeitenCushing IncidClaudia IrimieNoch keine Bewertungen
- Clinical Suspicion and Screening Tests for Cushing's SyndromeDokument9 SeitenClinical Suspicion and Screening Tests for Cushing's SyndromeHilalyNoch keine Bewertungen
- Adrenal MedullaDokument32 SeitenAdrenal MedullaAmanda MartinezNoch keine Bewertungen
- Adrenal Medulla and PheochromocytomaDokument32 SeitenAdrenal Medulla and PheochromocytomaAmanda MartinezNoch keine Bewertungen
- Cushing Syndrome: M.Sc. (N) 1 YearDokument30 SeitenCushing Syndrome: M.Sc. (N) 1 YearRanjana SharmaNoch keine Bewertungen
- Clinical Seminar on PheochromocytomaDokument9 SeitenClinical Seminar on PheochromocytomaParvathy JoshyNoch keine Bewertungen
- Probability Calculation OSPE Royal Free Hospital 2014Dokument29 SeitenProbability Calculation OSPE Royal Free Hospital 2014monday125Noch keine Bewertungen
- 6.23.08 Dancel Endocrine BD RevDokument32 Seiten6.23.08 Dancel Endocrine BD Revkhaled_71111Noch keine Bewertungen
- Disorders of The Adrenal Glands: Adrenal Cortical Adenoma and CarcinomaDokument9 SeitenDisorders of The Adrenal Glands: Adrenal Cortical Adenoma and Carcinomahussain AltaherNoch keine Bewertungen
- Adrenocortical HyperfunctionDokument132 SeitenAdrenocortical Hyperfunctionshobharamkrishna100% (2)
- Adrenal Gland Disorders: Cushing's, Primary Aldosteronism & MoreDokument9 SeitenAdrenal Gland Disorders: Cushing's, Primary Aldosteronism & Morehussain AltaherNoch keine Bewertungen
- Limits of DexamethasoneDokument17 SeitenLimits of DexamethasonesimonagaloiuNoch keine Bewertungen
- Adrenal Gland: Biochemistry by ZonesDokument12 SeitenAdrenal Gland: Biochemistry by ZonesWael AlkhatibNoch keine Bewertungen
- Case Report: Grace Y. Kim, MD Helen Anaedo, MD Sahar Nozad, MD Tipu Nazeer, MD Hassan Shawa, MDDokument6 SeitenCase Report: Grace Y. Kim, MD Helen Anaedo, MD Sahar Nozad, MD Tipu Nazeer, MD Hassan Shawa, MDPedro Gómez RNoch keine Bewertungen
- Macroprolactinemia Revisited A Study On 106 Patients JcemDokument8 SeitenMacroprolactinemia Revisited A Study On 106 Patients JcemPaulo Eduardo CampanaNoch keine Bewertungen
- The Cushing Syndrome: An Update On Diagnostic Tests: Determining CauseDokument12 SeitenThe Cushing Syndrome: An Update On Diagnostic Tests: Determining CauseRay Erick RamosNoch keine Bewertungen
- Uro2008, Vol.35, Issues 3, Minimally Invasive Genitourinary ProceduresDokument180 SeitenUro2008, Vol.35, Issues 3, Minimally Invasive Genitourinary ProcedureschuvanlaiNoch keine Bewertungen
- Case History 6.2: Box 6.13 Think of Unusual Causes of Hypertension, Especially in Younger PatientsDokument6 SeitenCase History 6.2: Box 6.13 Think of Unusual Causes of Hypertension, Especially in Younger PatientsDirgantari PademmeNoch keine Bewertungen
- Methimazole Drug Induced Agranulocytosis DefinitionDokument21 SeitenMethimazole Drug Induced Agranulocytosis DefinitionReyza ReyzaNoch keine Bewertungen
- Letter To The Editor Henoch-Schönlein Purpura in Adults: Clinics 2008 63 (2) :273-6Dokument4 SeitenLetter To The Editor Henoch-Schönlein Purpura in Adults: Clinics 2008 63 (2) :273-6donkeyendutNoch keine Bewertungen
- Approach To The PatientDokument11 SeitenApproach To The PatientNaya FarmsiBthreendy X'iNoch keine Bewertungen
- Case Hipertensi SekunderDokument8 SeitenCase Hipertensi Sekunderkintan utariNoch keine Bewertungen
- Endocrine QuizDokument34 SeitenEndocrine Quizakash kondapalliNoch keine Bewertungen
- Endocrine QuestionDokument110 SeitenEndocrine QuestionTofik MohammedNoch keine Bewertungen
- Androgen Excess in Women Experience With Over 1000Dokument10 SeitenAndrogen Excess in Women Experience With Over 1000Luisa Salazar VillegasNoch keine Bewertungen
- Successful Treatment of Myxedema Coma: A Case ReportDokument18 SeitenSuccessful Treatment of Myxedema Coma: A Case ReportElysabeth MargarethaNoch keine Bewertungen
- Unexpected Immunohistochemical Surprise of A Cushings Disease Macroadenoma Co-Expressing Acth and Prolactin: A Case ReportDokument5 SeitenUnexpected Immunohistochemical Surprise of A Cushings Disease Macroadenoma Co-Expressing Acth and Prolactin: A Case ReportIJAR JOURNALNoch keine Bewertungen
- Adrenal Gland Disorders ExplainedDokument48 SeitenAdrenal Gland Disorders ExplainedMubeenUrRehmanNoch keine Bewertungen
- Evaluation of Stress Hormones in Traumatic Brain Injury Patients With Gastrointestinal BleedingDokument7 SeitenEvaluation of Stress Hormones in Traumatic Brain Injury Patients With Gastrointestinal BleedingIvonne SoelionoNoch keine Bewertungen
- 2.06.08 Cushing's Syndrome McClune - 2Dokument24 Seiten2.06.08 Cushing's Syndrome McClune - 2IkbarardianNoch keine Bewertungen
- Cushing's Syndrome: Excess Cortisol and Its CausesDokument3 SeitenCushing's Syndrome: Excess Cortisol and Its CausesYoga KarsendaNoch keine Bewertungen
- Síndrome de Cushing. Maybe Not So Uncommon of An Endocrine DiseaseDokument10 SeitenSíndrome de Cushing. Maybe Not So Uncommon of An Endocrine DiseaseStephany Gutiérrez VargasNoch keine Bewertungen
- Ep171905 CRDokument6 SeitenEp171905 CRTantri SyahtiraNoch keine Bewertungen
- Treatment of Adrenalectomy Intraoperative Complications in A Patient With Adrenal Tumor Accompanied by Hypercortisol ManifestationDokument13 SeitenTreatment of Adrenalectomy Intraoperative Complications in A Patient With Adrenal Tumor Accompanied by Hypercortisol ManifestationRainbow DashieNoch keine Bewertungen
- Cushing Syndrome: (Hypercortisolism)Dokument7 SeitenCushing Syndrome: (Hypercortisolism)Daniela100% (1)
- PHEOCHROMOCYTOMADokument37 SeitenPHEOCHROMOCYTOMAYosi OktarinaNoch keine Bewertungen
- Cushing's SyndromeDokument35 SeitenCushing's SyndromeYoga KarsendaNoch keine Bewertungen
- Cushing's SyndromeDokument35 SeitenCushing's SyndromeShankara Pillai100% (3)
- DengueDokument10 SeitenDengueAlvaro Andres Flores JimenezNoch keine Bewertungen
- Arterioscler Thromb Vasc Biol 2002 Von Bahr 1129 35Dokument7 SeitenArterioscler Thromb Vasc Biol 2002 Von Bahr 1129 35widyaanggariniNoch keine Bewertungen
- Role of Tumour Marker in UrologyDokument5 SeitenRole of Tumour Marker in UrologyAung Ko HtetNoch keine Bewertungen
- SCE Practicaldiabetes - ElzohryDokument251 SeitenSCE Practicaldiabetes - ElzohryAE100% (1)
- Cushing DiseaseDokument24 SeitenCushing DiseaseSuci AlimaNoch keine Bewertungen
- 1213 0pdffileDokument5 Seiten1213 0pdffilecomedyvideo2220Noch keine Bewertungen
- Avs PDFDokument3 SeitenAvs PDFAmirah Farhanah AmiruddinNoch keine Bewertungen
- Malignant Giant Pheochromocytoma Case Report and Literature ReviewDokument8 SeitenMalignant Giant Pheochromocytoma Case Report and Literature ReviewannesasoumyanNoch keine Bewertungen
- 3f - 1 (Division of Task)Dokument33 Seiten3f - 1 (Division of Task)Trisha ArtosNoch keine Bewertungen
- AdrenalDokument19 SeitenAdrenalRumela Ganguly ChakrabortyNoch keine Bewertungen
- Cancer Res 1997 Zhuang 4682 6Dokument6 SeitenCancer Res 1997 Zhuang 4682 6Abdur Rachman Ba'abdullahNoch keine Bewertungen
- Primary Aldosteronism Caused by Unilateral Adrenal Hyperplasia: Rethinking The Accuracy of Imaging StudiesDokument5 SeitenPrimary Aldosteronism Caused by Unilateral Adrenal Hyperplasia: Rethinking The Accuracy of Imaging StudiesRonald DelosendoNoch keine Bewertungen
- Management of Cushing Syndrome in Children and Adolescents Experience of A Single Tertiary CentreDokument10 SeitenManagement of Cushing Syndrome in Children and Adolescents Experience of A Single Tertiary CentreTriratna FauziahNoch keine Bewertungen
- New Advances in Diagnosis and Treatment of Cushing SyndromeDokument12 SeitenNew Advances in Diagnosis and Treatment of Cushing SyndromeIka AnisaNoch keine Bewertungen
- Prognostic Value of Increased Soluble Thrornbornodulin and Increased Soluble E-Selectin in Ischaernic Heart DiseaseDokument6 SeitenPrognostic Value of Increased Soluble Thrornbornodulin and Increased Soluble E-Selectin in Ischaernic Heart DiseaseIoanna Bianca HNoch keine Bewertungen
- LEC 4 Adrenal HyperfunctionDokument39 SeitenLEC 4 Adrenal Hyperfunctionalhusien.abd2000Noch keine Bewertungen
- Adrenal Gland Disoreders 2Dokument14 SeitenAdrenal Gland Disoreders 2کشف FazilatNoch keine Bewertungen
- The Cleveland Clinic Manual of Dynamic Endocrine TestingVon EverandThe Cleveland Clinic Manual of Dynamic Endocrine TestingNoch keine Bewertungen
- Red Cotton Dress: By: Jelena MitićDokument4 SeitenRed Cotton Dress: By: Jelena MitićНиколина ГроздановскаNoch keine Bewertungen
- Graph Dynamic Linear Equations Student HandoutDokument6 SeitenGraph Dynamic Linear Equations Student HandoutНиколина ГроздановскаNoch keine Bewertungen
- Crochet BagsDokument25 SeitenCrochet Bagsformolici86% (7)
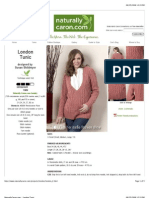
- London Tunic: Designed by Susan ShildmyerDokument5 SeitenLondon Tunic: Designed by Susan ShildmyerНиколина ГроздановскаNoch keine Bewertungen
- CROCHET - Amsterdam CoatDokument7 SeitenCROCHET - Amsterdam Coattritidief100% (2)
- Boobietraps YellowDokument3 SeitenBoobietraps YellowНиколина ГроздановскаNoch keine Bewertungen
- Aspira One Button TunicDokument3 SeitenAspira One Button TunicPilar R. Mendez100% (1)
- Bear Creek Coat: Designed Exclusively For Kraemer YarnsDokument2 SeitenBear Creek Coat: Designed Exclusively For Kraemer YarnsНиколина Гроздановска100% (1)
- Carmela LO34Dokument2 SeitenCarmela LO34Николина Гроздановска100% (1)
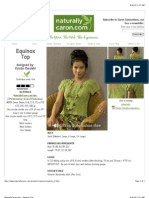
- EquinoxDokument3 SeitenEquinoxНиколина ГроздановскаNoch keine Bewertungen
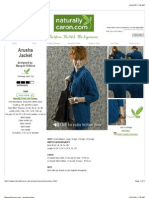
- ArushaDokument5 SeitenArushaНиколина ГроздановскаNoch keine Bewertungen
- Botanico NDokument3 SeitenBotanico Ncristina_marinescu_1Noch keine Bewertungen
- 7 Free Sweater PatternsDokument32 Seiten7 Free Sweater PatternsonutzamNoch keine Bewertungen
- Free Crochet Pattern Lion Brand Cotton-Ease (New) V-Stitch Wrap-Around SweaterDokument4 SeitenFree Crochet Pattern Lion Brand Cotton-Ease (New) V-Stitch Wrap-Around SweaterНиколина ГроздановскаNoch keine Bewertungen
- 0812 CM Tunisian5Dokument18 Seiten0812 CM Tunisian5Elvira KolutacNoch keine Bewertungen
- How To Be HealthyDokument8 SeitenHow To Be HealthyНиколина ГроздановскаNoch keine Bewertungen
- AdDokument5 SeitenAdНиколина ГроздановскаNoch keine Bewertungen
- IQ Boosters Success Sheet With PicturesDokument2 SeitenIQ Boosters Success Sheet With PicturesНиколина ГроздановскаNoch keine Bewertungen
- Yang - A New Trick To A Routine Procedure Taking The Fear Out of The Axillary Vein Stick Using The 35' Caudal View - Europace 2015Dokument4 SeitenYang - A New Trick To A Routine Procedure Taking The Fear Out of The Axillary Vein Stick Using The 35' Caudal View - Europace 2015Gabriel JoséNoch keine Bewertungen
- HeliotherapyDokument26 SeitenHeliotherapyalmont1759Noch keine Bewertungen
- Basic ECG Interpretation Review Questions Flashcards - QuizletDokument5 SeitenBasic ECG Interpretation Review Questions Flashcards - Quizletdina sharafNoch keine Bewertungen
- Heart Valve Disease Treatment Guide - Cleveland ClinicDokument12 SeitenHeart Valve Disease Treatment Guide - Cleveland ClinicGuillermo CenturionNoch keine Bewertungen
- Antihistamines PDFDokument47 SeitenAntihistamines PDFNoreak SokNoch keine Bewertungen
- Iontophoresis TENNIS ELBOW (Lateral Humeral Epicondylitis)Dokument2 SeitenIontophoresis TENNIS ELBOW (Lateral Humeral Epicondylitis)Ikre19Noch keine Bewertungen
- Antibiotics: Uses ActionDokument11 SeitenAntibiotics: Uses Actionammar amerNoch keine Bewertungen
- Book-Based Pathophysiology of Allergic and Non-Allergic RhinitisDokument2 SeitenBook-Based Pathophysiology of Allergic and Non-Allergic RhinitisJeraldine Corpuz PascualNoch keine Bewertungen
- TheTruth About Food Grade Hydrogen Peroxide H2O2 by James Paul RoguskiDokument86 SeitenTheTruth About Food Grade Hydrogen Peroxide H2O2 by James Paul RoguskiTheRazorsEdge80% (5)
- Medical TourismDokument55 SeitenMedical TourismLasik DelhiNoch keine Bewertungen
- NCMB 312 Rle MS Final Exam ReviewerDokument50 SeitenNCMB 312 Rle MS Final Exam Reviewer2-YA-4 ABEGAEL FERNANDEZNoch keine Bewertungen
- JaundiceDokument6 SeitenJaundiceNiaNoch keine Bewertungen
- 920 FullDokument17 Seiten920 FullHeru SigitNoch keine Bewertungen
- Oncology, Palliative Geriatric NursingDokument6 SeitenOncology, Palliative Geriatric Nursingminoshipeiris28Noch keine Bewertungen
- Promoting Maternal and Child HealthDokument2 SeitenPromoting Maternal and Child HealthShheeeeeshh100% (1)
- Rocket Seldinger Chest Drainage Set-UkDokument1 SeiteRocket Seldinger Chest Drainage Set-UktutagNoch keine Bewertungen
- OB Exam 1 Study GuideDokument23 SeitenOB Exam 1 Study GuideAlvin ManiagoNoch keine Bewertungen
- Cardiac Tumors: Dr. Asif WazirDokument20 SeitenCardiac Tumors: Dr. Asif WazirHillary BushnellNoch keine Bewertungen
- Pharmacology Intravenous Anesthetic Agents DissociativesDokument13 SeitenPharmacology Intravenous Anesthetic Agents DissociativesInnocent MwilaNoch keine Bewertungen
- Child Health FormDokument4 SeitenChild Health FormAaron Jay MondayaNoch keine Bewertungen
- Test Bank For Essentials of Pharmacology For Health Professions 7th Edition by WoodrowDokument9 SeitenTest Bank For Essentials of Pharmacology For Health Professions 7th Edition by WoodrowcandyblueNoch keine Bewertungen
- Question Preparation Exam2023-1Dokument350 SeitenQuestion Preparation Exam2023-1alicNoch keine Bewertungen
- London Core Review Course - (Thurs 18 May - Sun 21 May 2017) - TimetableDokument5 SeitenLondon Core Review Course - (Thurs 18 May - Sun 21 May 2017) - Timetablelondon_core_reviewNoch keine Bewertungen
- Scientific American Brasil - Abril 2020Dokument69 SeitenScientific American Brasil - Abril 2020rafaelNoch keine Bewertungen
- Ambulatory Blood Pressure MonitoringDokument26 SeitenAmbulatory Blood Pressure MonitoringnohazzNoch keine Bewertungen
- WHO Top 10 Causes of DeathDokument4 SeitenWHO Top 10 Causes of DeathMas IyoNoch keine Bewertungen
- Contribution of Intraoperative Electroencephalogram Suppression To Frailty-Associated Postoperative Delirium: Mediation Analysis of A Prospective Surgical CohortDokument9 SeitenContribution of Intraoperative Electroencephalogram Suppression To Frailty-Associated Postoperative Delirium: Mediation Analysis of A Prospective Surgical CohortSmith JaneNoch keine Bewertungen
- NEBULIZATIONDokument9 SeitenNEBULIZATIONSREEDEVI T SURESHNoch keine Bewertungen
- Icd 10Dokument229 SeitenIcd 10nilakarmilaNoch keine Bewertungen
- Week 2 - Elements of Physical Therapy ProcessDokument58 SeitenWeek 2 - Elements of Physical Therapy ProcessMicah Victoria Banes100% (1)