Das könnte Ihnen auch gefallen
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Von EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Bewertung: 4.5 von 5 Sternen4.5/5 (119)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaVon EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaBewertung: 4.5 von 5 Sternen4.5/5 (265)
- The Little Book of Hygge: Danish Secrets to Happy LivingVon EverandThe Little Book of Hygge: Danish Secrets to Happy LivingBewertung: 3.5 von 5 Sternen3.5/5 (399)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryVon EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryBewertung: 3.5 von 5 Sternen3.5/5 (231)
- Grit: The Power of Passion and PerseveranceVon EverandGrit: The Power of Passion and PerseveranceBewertung: 4 von 5 Sternen4/5 (587)
- Never Split the Difference: Negotiating As If Your Life Depended On ItVon EverandNever Split the Difference: Negotiating As If Your Life Depended On ItBewertung: 4.5 von 5 Sternen4.5/5 (838)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeVon EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeBewertung: 4 von 5 Sternen4/5 (5794)
- Team of Rivals: The Political Genius of Abraham LincolnVon EverandTeam of Rivals: The Political Genius of Abraham LincolnBewertung: 4.5 von 5 Sternen4.5/5 (234)
- Shoe Dog: A Memoir by the Creator of NikeVon EverandShoe Dog: A Memoir by the Creator of NikeBewertung: 4.5 von 5 Sternen4.5/5 (537)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyVon EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyBewertung: 3.5 von 5 Sternen3.5/5 (2219)
- The Emperor of All Maladies: A Biography of CancerVon EverandThe Emperor of All Maladies: A Biography of CancerBewertung: 4.5 von 5 Sternen4.5/5 (271)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreVon EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreBewertung: 4 von 5 Sternen4/5 (1090)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersVon EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersBewertung: 4.5 von 5 Sternen4.5/5 (344)
- Her Body and Other Parties: StoriesVon EverandHer Body and Other Parties: StoriesBewertung: 4 von 5 Sternen4/5 (821)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceVon EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceBewertung: 4 von 5 Sternen4/5 (894)
- I Could Easily FallDokument3 SeitenI Could Easily FallBenji100% (1)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureVon EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureBewertung: 4.5 von 5 Sternen4.5/5 (474)
- The Unwinding: An Inner History of the New AmericaVon EverandThe Unwinding: An Inner History of the New AmericaBewertung: 4 von 5 Sternen4/5 (45)
- Motivate! 2 End-Of-Term Test Standard: Units 1-3Dokument6 SeitenMotivate! 2 End-Of-Term Test Standard: Units 1-3Oum Vibol SatyaNoch keine Bewertungen
- The Yellow House: A Memoir (2019 National Book Award Winner)Von EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Bewertung: 4 von 5 Sternen4/5 (98)
- The Experience of God Being Consciousness BlissDokument376 SeitenThe Experience of God Being Consciousness BlissVivian Hyppolito100% (6)
- On Fire: The (Burning) Case for a Green New DealVon EverandOn Fire: The (Burning) Case for a Green New DealBewertung: 4 von 5 Sternen4/5 (73)
- Classical Music Forms ExplainedDokument11 SeitenClassical Music Forms Explainedcorinna_harrison100% (1)
- Identification and Management of Ambiguous GenitaliaDokument31 SeitenIdentification and Management of Ambiguous Genitaliateslimolakunleraji100% (1)
- Physical AssessmentDokument12 SeitenPhysical Assessmentteslimolakunleraji100% (1)
- Performance Requirements For Organic Coatings Applied To Under Hood and Chassis ComponentsDokument31 SeitenPerformance Requirements For Organic Coatings Applied To Under Hood and Chassis ComponentsIBR100% (2)
- Mar 2021Dokument2 SeitenMar 2021TanNoch keine Bewertungen
- Jamaica's Unemployment Aims, Causes and SolutionsDokument23 SeitenJamaica's Unemployment Aims, Causes and Solutionsnetzii300067% (3)
- Earth & Life Science Q1 Module 2 - DESIREE VICTORINODokument22 SeitenEarth & Life Science Q1 Module 2 - DESIREE VICTORINOJoshua A. Arabejo50% (4)
- Theory of Karma ExplainedDokument42 SeitenTheory of Karma ExplainedAKASH100% (1)
- Essay Sustainable Development GoalsDokument6 SeitenEssay Sustainable Development GoalsBima Dwi Nur Aziz100% (1)
- Top 10 Degrees For Emerging IndustriesDokument5 SeitenTop 10 Degrees For Emerging IndustriesteslimolakunlerajiNoch keine Bewertungen
- Femi Falana SANDokument1 SeiteFemi Falana SANteslimolakunlerajiNoch keine Bewertungen
- Femi Falana SANDokument1 SeiteFemi Falana SANteslimolakunlerajiNoch keine Bewertungen
- Tecno Pantom 6Dokument2 SeitenTecno Pantom 6teslimolakunlerajiNoch keine Bewertungen
- NigeriaDokument52 SeitenNigeriateslimolakunleraji100% (1)
- Top 10 Degrees For Emerging IndustriesDokument5 SeitenTop 10 Degrees For Emerging IndustriesteslimolakunlerajiNoch keine Bewertungen
- Getting Your MasterDokument3 SeitenGetting Your MasterteslimolakunlerajiNoch keine Bewertungen
- Obesity 1Dokument8 SeitenObesity 1teslimolakunlerajiNoch keine Bewertungen
- Interstitial FluidDokument2 SeitenInterstitial FluidteslimolakunlerajiNoch keine Bewertungen
- Abuth MDG PresentationDokument72 SeitenAbuth MDG PresentationteslimolakunlerajiNoch keine Bewertungen
- Attention Deficit Hyperactivity DisorderDokument21 SeitenAttention Deficit Hyperactivity DisorderteslimolakunlerajiNoch keine Bewertungen
- Control of Sickle Cell AmaemiaDokument9 SeitenControl of Sickle Cell AmaemiateslimolakunlerajiNoch keine Bewertungen
- MalariaDokument27 SeitenMalariaGaurav DharodNoch keine Bewertungen
- A Review On The Prevalence of Disabilities in ChildrenDokument11 SeitenA Review On The Prevalence of Disabilities in ChildrenteslimolakunlerajiNoch keine Bewertungen
- INDOMETACINDokument5 SeitenINDOMETACINteslimolakunlerajiNoch keine Bewertungen
- NeonatologyDokument4 SeitenNeonatologyteslimolakunleraji100% (1)
- LymphDokument3 SeitenLymphteslimolakunleraji100% (1)
- Classification & Clinical Features of LeprosyDokument41 SeitenClassification & Clinical Features of LeprosyteslimolakunlerajiNoch keine Bewertungen
- Glo Me Rulo NephritisDokument5 SeitenGlo Me Rulo NephritisteslimolakunlerajiNoch keine Bewertungen
- CancerDokument15 SeitenCancerteslimolakunlerajiNoch keine Bewertungen
- PneumoniaDokument8 SeitenPneumoniateslimolakunlerajiNoch keine Bewertungen
- HEMOPTYSISDokument3 SeitenHEMOPTYSISteslimolakunlerajiNoch keine Bewertungen
- Right-To-Left ShuntDokument16 SeitenRight-To-Left ShuntteslimolakunlerajiNoch keine Bewertungen
- Ebola VirusDokument5 SeitenEbola VirusteslimolakunlerajiNoch keine Bewertungen
- Scarlet FeverDokument12 SeitenScarlet FeverteslimolakunlerajiNoch keine Bewertungen
- VIRAL Haemorrhagic FeversDokument3 SeitenVIRAL Haemorrhagic FeversteslimolakunlerajiNoch keine Bewertungen
- ELEPHANTIASISDokument2 SeitenELEPHANTIASISteslimolakunlerajiNoch keine Bewertungen
- Nail ClubbingDokument2 SeitenNail ClubbingteslimolakunlerajiNoch keine Bewertungen
- First State of The Nation Address of ArroyoDokument9 SeitenFirst State of The Nation Address of ArroyoJennifer Sisperez Buraga-Waña LptNoch keine Bewertungen
- Endoplasmic ReticulumDokument4 SeitenEndoplasmic Reticulumnikki_fuentes_1100% (1)
- Fiegel Kutter Idriss PDFDokument1 SeiteFiegel Kutter Idriss PDFAvaNoch keine Bewertungen
- Development Proposal ReportDokument37 SeitenDevelopment Proposal ReportJean-Pierre RouxNoch keine Bewertungen
- Ramesh Dargond Shine Commerce Classes NotesDokument11 SeitenRamesh Dargond Shine Commerce Classes NotesRajath KumarNoch keine Bewertungen
- OutletsDokument226 SeitenOutletsPraveen Kumar Saini100% (1)
- Noise Blinking LED: Circuit Microphone Noise Warning SystemDokument1 SeiteNoise Blinking LED: Circuit Microphone Noise Warning Systemian jheferNoch keine Bewertungen
- Understanding key abdominal anatomy termsDokument125 SeitenUnderstanding key abdominal anatomy termscassandroskomplexNoch keine Bewertungen
- ENTH 311 Course Video ReflectionDokument2 SeitenENTH 311 Course Video ReflectionJeshua ItemNoch keine Bewertungen
- Compatibility Testing: Week 5Dokument33 SeitenCompatibility Testing: Week 5Bridgette100% (1)
- Alberta AwdNomineeDocs Case Circle BestMagazine NewTrailSpring2016Dokument35 SeitenAlberta AwdNomineeDocs Case Circle BestMagazine NewTrailSpring2016LucasNoch keine Bewertungen
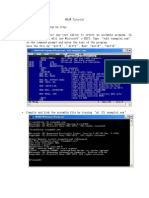
- MASM Tutorial PDFDokument10 SeitenMASM Tutorial PDFShashankDwivediNoch keine Bewertungen
- Food Processing & ClassificationDokument3 SeitenFood Processing & ClassificationAzrielle JaydeNoch keine Bewertungen
- C++ Project On Library Management by KCDokument53 SeitenC++ Project On Library Management by KCkeval71% (114)
- Reviewer in Intermediate Accounting IDokument9 SeitenReviewer in Intermediate Accounting ICzarhiena SantiagoNoch keine Bewertungen
- Unit-2 Fourier Series & Integral: 2130002 - Advanced Engineering MathematicsDokument143 SeitenUnit-2 Fourier Series & Integral: 2130002 - Advanced Engineering MathematicsDarji DhrutiNoch keine Bewertungen
- HDFC Bank's Organizational Profile and BackgroundDokument72 SeitenHDFC Bank's Organizational Profile and Backgroundrohitkh28Noch keine Bewertungen
- Situation AnalysisDokument94 SeitenSituation Analysisamirafateha100% (2)
- 150 Most Common Regular VerbsDokument4 Seiten150 Most Common Regular VerbsyairherreraNoch keine Bewertungen
- Chapter 018Dokument12 SeitenChapter 018api-281340024Noch keine Bewertungen