Das könnte Ihnen auch gefallen
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeVon EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeBewertung: 4 von 5 Sternen4/5 (5782)
- Shoe Dog: A Memoir by the Creator of NikeVon EverandShoe Dog: A Memoir by the Creator of NikeBewertung: 4.5 von 5 Sternen4.5/5 (537)
- The Little Book of Hygge: Danish Secrets to Happy LivingVon EverandThe Little Book of Hygge: Danish Secrets to Happy LivingBewertung: 3.5 von 5 Sternen3.5/5 (399)
- The Yellow House: A Memoir (2019 National Book Award Winner)Von EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Bewertung: 4 von 5 Sternen4/5 (98)
- On Fire: The (Burning) Case for a Green New DealVon EverandOn Fire: The (Burning) Case for a Green New DealBewertung: 4 von 5 Sternen4/5 (72)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceVon EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceBewertung: 4 von 5 Sternen4/5 (890)
- The Emperor of All Maladies: A Biography of CancerVon EverandThe Emperor of All Maladies: A Biography of CancerBewertung: 4.5 von 5 Sternen4.5/5 (271)
- Team of Rivals: The Political Genius of Abraham LincolnVon EverandTeam of Rivals: The Political Genius of Abraham LincolnBewertung: 4.5 von 5 Sternen4.5/5 (234)
- Never Split the Difference: Negotiating As If Your Life Depended On ItVon EverandNever Split the Difference: Negotiating As If Your Life Depended On ItBewertung: 4.5 von 5 Sternen4.5/5 (838)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureVon EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureBewertung: 4.5 von 5 Sternen4.5/5 (474)
- Grit: The Power of Passion and PerseveranceVon EverandGrit: The Power of Passion and PerseveranceBewertung: 4 von 5 Sternen4/5 (587)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryVon EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryBewertung: 3.5 von 5 Sternen3.5/5 (231)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaVon EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaBewertung: 4.5 von 5 Sternen4.5/5 (265)
- The Unwinding: An Inner History of the New AmericaVon EverandThe Unwinding: An Inner History of the New AmericaBewertung: 4 von 5 Sternen4/5 (45)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersVon EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersBewertung: 4.5 von 5 Sternen4.5/5 (344)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyVon EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyBewertung: 3.5 von 5 Sternen3.5/5 (2219)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreVon EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreBewertung: 4 von 5 Sternen4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Von EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Bewertung: 4.5 von 5 Sternen4.5/5 (119)
- Her Body and Other Parties: StoriesVon EverandHer Body and Other Parties: StoriesBewertung: 4 von 5 Sternen4/5 (821)
- Regulation of RespirationDokument46 SeitenRegulation of Respirationnirilib89% (9)
- Comprehensive List of Collective NounsDokument5 SeitenComprehensive List of Collective NounsRItuparnaNoch keine Bewertungen
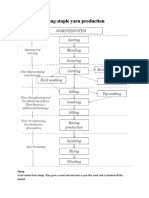
- Sequence of Process in Worsted SpinningDokument4 SeitenSequence of Process in Worsted SpinningArunraj Arumugam100% (4)
- InvertibrateDokument10 SeitenInvertibrateSerdar AgidNoch keine Bewertungen
- This Is It - Myoma Case PresentationDokument93 SeitenThis Is It - Myoma Case Presentationapi-1976296791% (11)
- D&D 5e Random Character Generator 8.0Dokument2.346 SeitenD&D 5e Random Character Generator 8.0aallievi67% (3)
- Revision Notes.Dokument32 SeitenRevision Notes.drhaj70% (1)
- Dragon and Tiger Qigong 161 180Dokument20 SeitenDragon and Tiger Qigong 161 180mamun31Noch keine Bewertungen
- Anatomy IDokument2 SeitenAnatomy ISatria Panca KartaNoch keine Bewertungen
- Abbe Faria (2014) A Compendium of Articles and Photographs PDFDokument275 SeitenAbbe Faria (2014) A Compendium of Articles and Photographs PDFAmitranjan Basu100% (1)
- MMnewDokument19 SeitenMMnewneocrust123Noch keine Bewertungen
- Veterinary Epidemiology (VEP-411)Dokument15 SeitenVeterinary Epidemiology (VEP-411)Vijay Kumar Anumolu100% (1)
- SlatkovodneribewebDokument105 SeitenSlatkovodneribewebJasmin HabibovicNoch keine Bewertungen
- Chapter 12 VocabDokument2 SeitenChapter 12 VocabwdsfNoch keine Bewertungen
- Brain Stem AnatomiDokument55 SeitenBrain Stem AnatomiNor Ubudiah SetiNoch keine Bewertungen
- We Ate The Children LastDokument3 SeitenWe Ate The Children LastjeniouslyNoch keine Bewertungen
- Rivero 1991. New Ecuadorean Colostethus (Amphibia, Dendrobatidae) in The Collection of The National Museum of Natural History, Smithsonian InstitutionDokument16 SeitenRivero 1991. New Ecuadorean Colostethus (Amphibia, Dendrobatidae) in The Collection of The National Museum of Natural History, Smithsonian InstitutionNicole AcosVas0% (1)
- Pet Size o Meter Guinea PigDokument2 SeitenPet Size o Meter Guinea PigLiong KimNoch keine Bewertungen
- Minor ailments of the postpartum periodDokument4 SeitenMinor ailments of the postpartum periodHarish LabanaNoch keine Bewertungen
- Papular UrticariaDokument10 SeitenPapular UrticariaNuri Sakina SuhartoNoch keine Bewertungen
- THE World of Naria: Animals Form NarniaDokument3 SeitenTHE World of Naria: Animals Form NarniaDragomir RaresNoch keine Bewertungen
- Friedman and Daeshler 2006 Late Devonian (Famennian) Lungfishes From The Catskill Formation of Pennsylvania, UsaDokument17 SeitenFriedman and Daeshler 2006 Late Devonian (Famennian) Lungfishes From The Catskill Formation of Pennsylvania, UsaFernando Arthur GregolNoch keine Bewertungen
- Principles of Spine Trauma and Spinal Deformities PDFDokument34 SeitenPrinciples of Spine Trauma and Spinal Deformities PDFVirlan Vasile CatalinNoch keine Bewertungen
- General Recommendation On ImmunizationDokument19 SeitenGeneral Recommendation On ImmunizationRickyNoviantoNoch keine Bewertungen
- Pa Auk The Chariot To Nibbana Part IIDokument32 SeitenPa Auk The Chariot To Nibbana Part IItjendra100% (1)
- General Insect Physiology 3344Dokument14 SeitenGeneral Insect Physiology 3344Shawn BlueNoch keine Bewertungen
- The Ant and The GrasshopperDokument12 SeitenThe Ant and The Grasshoppermarilou sorianoNoch keine Bewertungen
- 4 Feb - Reading Test 1 (SMA)Dokument4 Seiten4 Feb - Reading Test 1 (SMA)Lopian SilabanNoch keine Bewertungen
- Anatomy & Physiology With Pathophysiology Lecture (Midterms)Dokument4 SeitenAnatomy & Physiology With Pathophysiology Lecture (Midterms)Millen ArenasNoch keine Bewertungen
- The Fortune of Kali YugaDokument2 SeitenThe Fortune of Kali Yugayasaswy100% (1)