Das könnte Ihnen auch gefallen
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Von EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Bewertung: 4.5 von 5 Sternen4.5/5 (121)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryVon EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryBewertung: 3.5 von 5 Sternen3.5/5 (231)
- Grit: The Power of Passion and PerseveranceVon EverandGrit: The Power of Passion and PerseveranceBewertung: 4 von 5 Sternen4/5 (588)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaVon EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaBewertung: 4.5 von 5 Sternen4.5/5 (266)
- Never Split the Difference: Negotiating As If Your Life Depended On ItVon EverandNever Split the Difference: Negotiating As If Your Life Depended On ItBewertung: 4.5 von 5 Sternen4.5/5 (838)
- The Emperor of All Maladies: A Biography of CancerVon EverandThe Emperor of All Maladies: A Biography of CancerBewertung: 4.5 von 5 Sternen4.5/5 (271)
- The Little Book of Hygge: Danish Secrets to Happy LivingVon EverandThe Little Book of Hygge: Danish Secrets to Happy LivingBewertung: 3.5 von 5 Sternen3.5/5 (400)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeVon EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeBewertung: 4 von 5 Sternen4/5 (5794)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyVon EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyBewertung: 3.5 von 5 Sternen3.5/5 (2259)
- Shoe Dog: A Memoir by the Creator of NikeVon EverandShoe Dog: A Memoir by the Creator of NikeBewertung: 4.5 von 5 Sternen4.5/5 (537)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreVon EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreBewertung: 4 von 5 Sternen4/5 (1090)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersVon EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersBewertung: 4.5 von 5 Sternen4.5/5 (345)
- Team of Rivals: The Political Genius of Abraham LincolnVon EverandTeam of Rivals: The Political Genius of Abraham LincolnBewertung: 4.5 von 5 Sternen4.5/5 (234)
- Her Body and Other Parties: StoriesVon EverandHer Body and Other Parties: StoriesBewertung: 4 von 5 Sternen4/5 (821)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceVon EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceBewertung: 4 von 5 Sternen4/5 (895)
- The Unwinding: An Inner History of the New AmericaVon EverandThe Unwinding: An Inner History of the New AmericaBewertung: 4 von 5 Sternen4/5 (45)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureVon EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureBewertung: 4.5 von 5 Sternen4.5/5 (474)
- On Fire: The (Burning) Case for a Green New DealVon EverandOn Fire: The (Burning) Case for a Green New DealBewertung: 4 von 5 Sternen4/5 (74)
- The Yellow House: A Memoir (2019 National Book Award Winner)Von EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Bewertung: 4 von 5 Sternen4/5 (98)
- AERZEN - DTR - BOGE - Osuszacze Medyczne Serrii DASZ-P 1-2... 14-2Dokument68 SeitenAERZEN - DTR - BOGE - Osuszacze Medyczne Serrii DASZ-P 1-2... 14-2PiotrNoch keine Bewertungen
- Studying Gold Ores: Mineralogy, Cyanidation, Toxicity and Environmental IssuesDokument34 SeitenStudying Gold Ores: Mineralogy, Cyanidation, Toxicity and Environmental IssuesFitria Putri DwiNoch keine Bewertungen
- Disperse Systems. The Methods of Preparing of Colloidal Solutions. Their PropertiesDokument33 SeitenDisperse Systems. The Methods of Preparing of Colloidal Solutions. Their Propertieslsueyin100% (1)
- 3 Unit OperationsDokument36 Seiten3 Unit OperationsAkanksha SrivastavaNoch keine Bewertungen
- CO2 Adsorption Process Simulation in ASPEN HysysDokument5 SeitenCO2 Adsorption Process Simulation in ASPEN HysysHussseinmubarkNoch keine Bewertungen
- Chemical Reactions of Copper and Percent YieldDokument8 SeitenChemical Reactions of Copper and Percent Yieldlsueyin0% (1)
- Zeolites and Zeolite Like Material in Industrial CatalysisDokument33 SeitenZeolites and Zeolite Like Material in Industrial CatalysislsueyinNoch keine Bewertungen
- FullerenesDokument411 SeitenFullereneslsueyinNoch keine Bewertungen
- Towards The Rational Synthesis of ZeolitesDokument0 SeitenTowards The Rational Synthesis of ZeoliteslsueyinNoch keine Bewertungen
- Organic Photochemistry: Prof. Dr. Burkhard König, Institut Für Organische Chemie, Universität RegensburgDokument38 SeitenOrganic Photochemistry: Prof. Dr. Burkhard König, Institut Für Organische Chemie, Universität RegensburglsueyinNoch keine Bewertungen
- Foodweb KeyDokument3 SeitenFoodweb KeylsueyinNoch keine Bewertungen
- C++ ExerciseDokument49 SeitenC++ ExerciselsueyinNoch keine Bewertungen
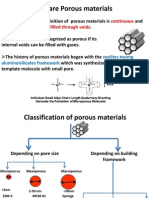
- What Are Porous MaterialsDokument33 SeitenWhat Are Porous MaterialslsueyinNoch keine Bewertungen
- Ion-Selective Electrode Determination of Fluoride Ion: Chemistry 321L ManualDokument5 SeitenIon-Selective Electrode Determination of Fluoride Ion: Chemistry 321L Manuallsueyin100% (1)
- ERT 313 Bioseparation Engineering Adsorption: Prepared By: Miss Hairul Nazirah Abdul HalimDokument25 SeitenERT 313 Bioseparation Engineering Adsorption: Prepared By: Miss Hairul Nazirah Abdul HalimlsueyinNoch keine Bewertungen
- Enzyme Kinetics and Catalysis IIDokument36 SeitenEnzyme Kinetics and Catalysis IIlsueyinNoch keine Bewertungen
- Einstein's Theory of Gravitation: Gravitational WavesDokument9 SeitenEinstein's Theory of Gravitation: Gravitational WaveslsueyinNoch keine Bewertungen
- Potentiometry: Suggested Problems From S.H.C.Dokument11 SeitenPotentiometry: Suggested Problems From S.H.C.lsueyinNoch keine Bewertungen
- Preparation of CatalystsDokument44 SeitenPreparation of CatalystslsueyinNoch keine Bewertungen
- Symmetry. Point Groups and Character Tables I, Symmetry Operations and Their Importance For Chemical ProblemsDokument7 SeitenSymmetry. Point Groups and Character Tables I, Symmetry Operations and Their Importance For Chemical ProblemslsueyinNoch keine Bewertungen
- MOF NoteDokument61 SeitenMOF NotelsueyinNoch keine Bewertungen
- FTIR IntroductionDokument8 SeitenFTIR Introductiontimtailieu2010Noch keine Bewertungen
- Persuasive Essay Sample Paper: Name - DateDokument1 SeitePersuasive Essay Sample Paper: Name - DatelsueyinNoch keine Bewertungen
- Sample Essay Exam ExerciseDokument2 SeitenSample Essay Exam ExerciselsueyinNoch keine Bewertungen
- Sample Passing Regents' Essays: 3/2 ModelDokument2 SeitenSample Passing Regents' Essays: 3/2 ModellsueyinNoch keine Bewertungen
- Simultaneous Determination of Copper and Iron in Automotive Gasoline by X Ray Fluorescence After Pre Concentration On Cellulose Paper 2007 TalantaDokument4 SeitenSimultaneous Determination of Copper and Iron in Automotive Gasoline by X Ray Fluorescence After Pre Concentration On Cellulose Paper 2007 TalantaPancho Agustín Miranda SantibáñezNoch keine Bewertungen
- PLC For The Pressure Swing Adsorption (PSA) SystemDokument4 SeitenPLC For The Pressure Swing Adsorption (PSA) SystempintaratNoch keine Bewertungen
- Removal of Hydrocarbons From Waste Waters by ZeoliteDokument84 SeitenRemoval of Hydrocarbons From Waste Waters by Zeoliteleila200quartzNoch keine Bewertungen
- 6 Simulation of An Adsorption Solar Cooling SystemDokument8 Seiten6 Simulation of An Adsorption Solar Cooling SystemBranislavPetrovicNoch keine Bewertungen
- Self-Assembled Mono Layers (Sams)Dokument10 SeitenSelf-Assembled Mono Layers (Sams)iskander_medNoch keine Bewertungen
- Mto MCQDokument51 SeitenMto MCQPrajwal ChavanNoch keine Bewertungen
- Coke Formation in Zeolite ZSM-5Dokument10 SeitenCoke Formation in Zeolite ZSM-5Neil MilestoneNoch keine Bewertungen
- NPTL 2Dokument254 SeitenNPTL 2vigeshNoch keine Bewertungen
- Adsorcion, BioadsorbentesDokument9 SeitenAdsorcion, BioadsorbentesCinthia Judith Romero CervantesNoch keine Bewertungen
- Adsorption Equil Principles - 483Dokument28 SeitenAdsorption Equil Principles - 483MarwahNoch keine Bewertungen
- Catalysis Communications: Rosilda Selvin, Hsiu-Ling Hsu, Tze-Min HerDokument4 SeitenCatalysis Communications: Rosilda Selvin, Hsiu-Ling Hsu, Tze-Min Herrommy agurto palaciosNoch keine Bewertungen
- Kinetics OverviewDokument128 SeitenKinetics OverviewDr. Srinivas MandavaNoch keine Bewertungen
- Hangzhou Chenrui Air Separator Installation Manufacture CO., LTDDokument10 SeitenHangzhou Chenrui Air Separator Installation Manufacture CO., LTDTonyNoch keine Bewertungen
- Optimization of Electrocoagulation (EC) Process For The Purification of Water From 2,4-Dichlorophenoxyacetic Acid (2,4-D) Using Sacrificial AnodesDokument24 SeitenOptimization of Electrocoagulation (EC) Process For The Purification of Water From 2,4-Dichlorophenoxyacetic Acid (2,4-D) Using Sacrificial AnodesANGIE ARLETTE HIDALGO APAZANoch keine Bewertungen
- A Study of The Deactivation of Low Loading Ni:Al2O3 Steam Reforming Catalyst by TetrahydrothiopheneDokument7 SeitenA Study of The Deactivation of Low Loading Ni:Al2O3 Steam Reforming Catalyst by TetrahydrothiopheneVương Duy NghiêmNoch keine Bewertungen
- 2018-MCM 48 Encapsulated With Reduced Graphene Oxide - Journal of Molecular Liquids-Akpotu PDFDokument10 Seiten2018-MCM 48 Encapsulated With Reduced Graphene Oxide - Journal of Molecular Liquids-Akpotu PDFDanCosminNoch keine Bewertungen
- Full Documentation MTX+ MAJ 260210Dokument212 SeitenFull Documentation MTX+ MAJ 260210m_1_3100% (4)
- Foam Fractionation ApplicationsDokument13 SeitenFoam Fractionation ApplicationsHaile GetachewNoch keine Bewertungen
- Carbon 04 00062 PDFDokument20 SeitenCarbon 04 00062 PDFHartinie MNoch keine Bewertungen
- Use of A New Size-Weighted Combining Rule To Predict Adsorption in Siliceous ZeolitesDokument17 SeitenUse of A New Size-Weighted Combining Rule To Predict Adsorption in Siliceous Zeolitessarvelos1Noch keine Bewertungen
- Samson ValvesDokument16 SeitenSamson ValvesjimmiilongNoch keine Bewertungen
- DFT Tutorial MatSQ Mosab BanisalmanDokument47 SeitenDFT Tutorial MatSQ Mosab BanisalmanSaliya Ranashigha BandaraNoch keine Bewertungen
- Materials: Drying in TraysDokument10 SeitenMaterials: Drying in TraysMira FazziraNoch keine Bewertungen
- UntitledDokument60 SeitenUntitledJatin ParajiyaNoch keine Bewertungen
- Adsorption and Desorption ProcessesDokument19 SeitenAdsorption and Desorption ProcessesĐạt DokonNoch keine Bewertungen
- General Characteristics of Thermally Cycled Tin Oxide Gas Sensors 1989Dokument10 SeitenGeneral Characteristics of Thermally Cycled Tin Oxide Gas Sensors 1989Koit MauringNoch keine Bewertungen