Das könnte Ihnen auch gefallen
- Preparation of Plasmid DNA by Alkaline Lysis With SDS MinipreparationDokument2 SeitenPreparation of Plasmid DNA by Alkaline Lysis With SDS Minipreparationstevensb055100% (1)
- Mini PrepDokument6 SeitenMini PrepWilson GomargaNoch keine Bewertungen
- Isolation of Plasmid DnaDokument6 SeitenIsolation of Plasmid Dnavictor0% (1)
- EXP5CHEM26Dokument12 SeitenEXP5CHEM26Albert Romano ObisNoch keine Bewertungen
- Estimation of DNADokument1 SeiteEstimation of DNATjcbt BiosciencesNoch keine Bewertungen
- Extraction of Genomic DNA: G.Umamaheswaran PH.D Scholar JipmerDokument24 SeitenExtraction of Genomic DNA: G.Umamaheswaran PH.D Scholar Jipmerpharmaguy111Noch keine Bewertungen
- Isolation of DNA Extraction From Plant TissueDokument6 SeitenIsolation of DNA Extraction From Plant Tissueanura7Noch keine Bewertungen
- Dna Purification and Extraction Practical ReportDokument8 SeitenDna Purification and Extraction Practical ReportAnselmo ManishaNoch keine Bewertungen
- Cloning VectorsDokument19 SeitenCloning VectorsvmshanesNoch keine Bewertungen
- Experiment - 2: Plasmid DNA Isolation Using Alkaline Lysis MethodDokument14 SeitenExperiment - 2: Plasmid DNA Isolation Using Alkaline Lysis MethodVineet Kumar ThakurNoch keine Bewertungen
- Enzyme Purification NotesDokument23 SeitenEnzyme Purification NotesMadan Rajan100% (1)
- DNA Isolation, Restriction, Visualitation, and QuantificationDokument20 SeitenDNA Isolation, Restriction, Visualitation, and QuantificationSonianto kuddi100% (5)
- Purification of EnzymeDokument29 SeitenPurification of Enzymeplastioid4079Noch keine Bewertungen
- Western Blotting MicrsoftDokument45 SeitenWestern Blotting MicrsoftSoNu de Bond100% (2)
- Biotecnology and Its ApplicationDokument87 SeitenBiotecnology and Its ApplicationRangaswamyBiligiraiah100% (1)
- BCH 314 Tutorial 1 SolutionsDokument9 SeitenBCH 314 Tutorial 1 SolutionsvictorNoch keine Bewertungen
- Isolation of Genomic DNA from MycobacteriaDokument2 SeitenIsolation of Genomic DNA from MycobacteriaGuhan KANoch keine Bewertungen
- Preparation of Plasmid DNA by Alkaline Lysis With SDSDokument5 SeitenPreparation of Plasmid DNA by Alkaline Lysis With SDStanjent50% (2)
- Lysis of Plasmidian CellDokument3 SeitenLysis of Plasmidian Cellodile1987Noch keine Bewertungen
- Isolation, Purification, and Characterization of Serratiopeptidase Enzyme From Serratia MarcescensDokument6 SeitenIsolation, Purification, and Characterization of Serratiopeptidase Enzyme From Serratia MarcescensInternational Journal of Innovative Science and Research TechnologyNoch keine Bewertungen
- Restriction Digestion of Plasmid DNADokument22 SeitenRestriction Digestion of Plasmid DNAMichelle100% (4)
- In-Depth Steps Towards Nucleic Acid and Protein SynthesisDokument21 SeitenIn-Depth Steps Towards Nucleic Acid and Protein SynthesisGbenga AjaniNoch keine Bewertungen
- Question 1 (37 Marks) : Biochemistry 3 BCH 314Dokument4 SeitenQuestion 1 (37 Marks) : Biochemistry 3 BCH 314victorNoch keine Bewertungen
- ABT 227 - Course Outline Introduction To Molecular Biology (3335)Dokument1 SeiteABT 227 - Course Outline Introduction To Molecular Biology (3335)justevansiNoch keine Bewertungen
- CentrifugationDokument20 SeitenCentrifugationMuhammad Asif ShaheenNoch keine Bewertungen
- Western BlottingDokument13 SeitenWestern BlottingAshfaq Fazal100% (1)
- DNA Manipulative EnzymesDokument17 SeitenDNA Manipulative EnzymesZain Ul AbedienNoch keine Bewertungen
- Principles of Cell Culture: A Brief History and OverviewDokument20 SeitenPrinciples of Cell Culture: A Brief History and OverviewMicah Marie Cao AngeliasNoch keine Bewertungen
- cDNA Libraries and Gene CloningDokument8 SeitencDNA Libraries and Gene CloningRoberto RomeroNoch keine Bewertungen
- Restriction Enzymes PDFDokument7 SeitenRestriction Enzymes PDFmanoj_rkl_07Noch keine Bewertungen
- Isolation of Genomic DNADokument8 SeitenIsolation of Genomic DNAMadhuri HarshaNoch keine Bewertungen
- VectorsDokument6 SeitenVectorsAssad MustafaNoch keine Bewertungen
- Recombinant DNA Technology: Dr. P. Balaji Head in Biotechnology MGR College, HosurDokument78 SeitenRecombinant DNA Technology: Dr. P. Balaji Head in Biotechnology MGR College, HosurBalaji Paulraj100% (1)
- M.Prasad Naidu MSC Medical Biochemistry, Ph.D.Research ScholarDokument39 SeitenM.Prasad Naidu MSC Medical Biochemistry, Ph.D.Research ScholarM.PRASAD NAIDUNoch keine Bewertungen
- RNA PolymeraseDokument14 SeitenRNA PolymeraseKanaka lata SorenNoch keine Bewertungen
- DNA ExtractionDokument5 SeitenDNA ExtractionDana Porter100% (1)
- Plant Biotechnology NotesDokument16 SeitenPlant Biotechnology NotesAnanya Singh100% (1)
- Selection of Recombinant ClonesDokument2 SeitenSelection of Recombinant ClonesDeepika KVNoch keine Bewertungen
- Cloning Dolly & MicromanipulationDokument29 SeitenCloning Dolly & Micromanipulationnitinyadav16Noch keine Bewertungen
- PlasmidsDokument53 SeitenPlasmidsPrerana SikarwarNoch keine Bewertungen
- Southern Blotting Techniques ExplainedDokument50 SeitenSouthern Blotting Techniques ExplainedSandeep Otari100% (1)
- Nucleic Acids As Genetic Information CarriersDokument32 SeitenNucleic Acids As Genetic Information Carriersstevensb055100% (4)
- Estimation of Citric Acid From Aspergillus SPDokument4 SeitenEstimation of Citric Acid From Aspergillus SPDinithiDahanayake100% (3)
- Production of Citric Acid by Aspergillus nigerDokument9 SeitenProduction of Citric Acid by Aspergillus nigerNikitaNoch keine Bewertungen
- Restriction Enzyme Digestion AnalysisDokument6 SeitenRestriction Enzyme Digestion AnalysisLloaana 12Noch keine Bewertungen
- Cloning VectorsDokument5 SeitenCloning VectorsMilo Kai HeeNoch keine Bewertungen
- Restriction Enzyme DigestionDokument5 SeitenRestriction Enzyme DigestionAqsa ImtiazNoch keine Bewertungen
- Determine Iron Content Using Permanganometric TitrationDokument13 SeitenDetermine Iron Content Using Permanganometric TitrationNahzim RahmatNoch keine Bewertungen
- Strain ImprovementDokument15 SeitenStrain ImprovementAlanChevalNoch keine Bewertungen
- Plasmid LabDokument10 SeitenPlasmid LabAhmed J AlhindaweNoch keine Bewertungen
- Bacterial GeneticsDokument9 SeitenBacterial GeneticsExamville.comNoch keine Bewertungen
- (Chem 102.2) Polymerase Chain ReactionDokument19 Seiten(Chem 102.2) Polymerase Chain ReactionRalph John UgalinoNoch keine Bewertungen
- Food Biotechnology (ORGANIC ACIDS, ENZYMES, FOOD ADDITIVES)Dokument61 SeitenFood Biotechnology (ORGANIC ACIDS, ENZYMES, FOOD ADDITIVES)Dumisani Nguni100% (1)
- PCR PDFDokument43 SeitenPCR PDFAmirNoch keine Bewertungen
- DNA Sequencing Methods ExplainedDokument21 SeitenDNA Sequencing Methods ExplainedAsfoor gake1Noch keine Bewertungen
- Substitution MatrixDokument10 SeitenSubstitution MatrixRashmi DhimanNoch keine Bewertungen
- Cloning VectorsDokument5 SeitenCloning Vectorssashaikh1213Noch keine Bewertungen
- Linkage and crossing over: Genetic recombination between genes on homologous chromosomesDokument34 SeitenLinkage and crossing over: Genetic recombination between genes on homologous chromosomesDhungana Surya RdNoch keine Bewertungen
- Decision Making TechniquesDokument6 SeitenDecision Making TechniquesKhandoker FaisalNoch keine Bewertungen
- Create Offset in MapinfoDokument4 SeitenCreate Offset in MapinfoKhandoker FaisalNoch keine Bewertungen
- Calculating Time Value of Money and Discounted Cash FlowDokument27 SeitenCalculating Time Value of Money and Discounted Cash FlowKhandoker FaisalNoch keine Bewertungen
- Basic Project Management On PMI Framework 2nd RelDokument65 SeitenBasic Project Management On PMI Framework 2nd RelKhandoker FaisalNoch keine Bewertungen
- Lecture4 Intro To WCDMA PDFDokument43 SeitenLecture4 Intro To WCDMA PDFA Chaerunisa Utami PutriNoch keine Bewertungen
- BSC6900 Parameter NamesDokument73 SeitenBSC6900 Parameter NamesJahid MasudNoch keine Bewertungen
- BSS Call PhasesDokument15 SeitenBSS Call PhasesKhandoker FaisalNoch keine Bewertungen
- IMPOTRANT Alcatel Parameters B7.2 0062 17aDokument606 SeitenIMPOTRANT Alcatel Parameters B7.2 0062 17aKhandoker FaisalNoch keine Bewertungen
- Rayleigh ChannelDokument12 SeitenRayleigh Channelshubham12Noch keine Bewertungen
- General Installation Practices: Section EDokument26 SeitenGeneral Installation Practices: Section EMohsen ArabifardNoch keine Bewertungen
- How To Make Black PowderDokument7 SeitenHow To Make Black Powder8mhno100% (1)
- Powder Metallurgy Process and ApplicationsDokument32 SeitenPowder Metallurgy Process and ApplicationsChandan KumarNoch keine Bewertungen
- Advion MSDSDokument6 SeitenAdvion MSDSmoespestcontrol_mnNoch keine Bewertungen
- Week No.2 - CLO-1 Size ReductionDokument18 SeitenWeek No.2 - CLO-1 Size ReductionkhalifaNoch keine Bewertungen
- Ganoderma laccase optimizationDokument9 SeitenGanoderma laccase optimizationRajeshKumarNoch keine Bewertungen
- Tubular Processing of The Glomerular FiltrateDokument8 SeitenTubular Processing of The Glomerular FiltrateOsama MohamedNoch keine Bewertungen
- 11 Numerical AnalysisDokument9 Seiten11 Numerical Analysisعزالدين حسنNoch keine Bewertungen
- The Chemistry of Heterocycles Structure, Reactions, Syntheses, and ApplicationsDokument571 SeitenThe Chemistry of Heterocycles Structure, Reactions, Syntheses, and Applications17.Hồ Nguyên Khang100% (1)
- 12 Amadi TEOP Master-Ed2013-14 PDFDokument25 Seiten12 Amadi TEOP Master-Ed2013-14 PDFluis_seczonNoch keine Bewertungen
- Hysys 8.8 - ManualDokument606 SeitenHysys 8.8 - ManualCarlos Vaz88% (8)
- Reservoir Drive Mechanisms PDFDokument28 SeitenReservoir Drive Mechanisms PDFWassef MB100% (1)
- Powder Metallurgy: Crushing and Milling Method To Form PowderDokument12 SeitenPowder Metallurgy: Crushing and Milling Method To Form Powder050678Noch keine Bewertungen
- Concrete Soil Identification GuideDokument3 SeitenConcrete Soil Identification GuideKangNoch keine Bewertungen
- Balancing Redox Reactions Worksheets 1 & 2 (With Answers) PDFDokument2 SeitenBalancing Redox Reactions Worksheets 1 & 2 (With Answers) PDFMohamed MeeranNoch keine Bewertungen
- Ferritic and Martensitic Casting Materials SpecificationsDokument2 SeitenFerritic and Martensitic Casting Materials SpecificationsSinan YıldızNoch keine Bewertungen
- Shell Momentum Balance 1Dokument16 SeitenShell Momentum Balance 1Kevwe Macaulay -GbogidiNoch keine Bewertungen
- MSDS Polyken 1027 PrimerDokument7 SeitenMSDS Polyken 1027 PrimerPungkas NisworoNoch keine Bewertungen
- Phys 1Dokument5 SeitenPhys 1Sandra Phan50% (2)
- Onion Cell Structure Under MicroscopeDokument2 SeitenOnion Cell Structure Under MicroscopeAnirudh100% (1)
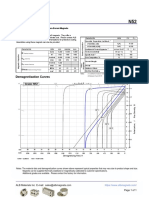
- N52 Grade Neodymium Magnets DataDokument1 SeiteN52 Grade Neodymium Magnets DataSteve HsuNoch keine Bewertungen
- Effectiveness of Liquid Oxygen BleachDokument4 SeitenEffectiveness of Liquid Oxygen BleachSingh GurleenNoch keine Bewertungen
- 31.PEAK Depressurization RATEDokument1 Seite31.PEAK Depressurization RATEDILIP MATALNoch keine Bewertungen
- A Simplified Method For The Cultivation of Extreme Anaerobic Archaea Based SULFIDE 2000 !!!!Dokument6 SeitenA Simplified Method For The Cultivation of Extreme Anaerobic Archaea Based SULFIDE 2000 !!!!Vera Brok-VolchanskayaNoch keine Bewertungen
- Vragen Fasediagrammen Zuivere Componenten PDFDokument3 SeitenVragen Fasediagrammen Zuivere Componenten PDFbilberNoch keine Bewertungen
- Quantum-Mechanical Aspects of The L. Pauling's Resonance Theory.Dokument4 SeitenQuantum-Mechanical Aspects of The L. Pauling's Resonance Theory.Bezverkhniy VolodymyrNoch keine Bewertungen
- Chapter 7 PDFDokument36 SeitenChapter 7 PDFRbtl BañosNoch keine Bewertungen
- Jakap Lace Pvt. LTD.: Material Test CertificateDokument1 SeiteJakap Lace Pvt. LTD.: Material Test CertificateMechtek LabNoch keine Bewertungen
- Vortex Quantum SeriesDokument34 SeitenVortex Quantum SeriesmiguelcNoch keine Bewertungen
- On Water Cycle With Unlocking of DifficultiesDokument38 SeitenOn Water Cycle With Unlocking of DifficultiesShirly Basilio100% (1)