Das könnte Ihnen auch gefallen
- PYS 3.1 DharanaDokument2 SeitenPYS 3.1 Dharanasreeraj.guruvayoor50% (2)
- Matha Amrithananda MayiDokument4 SeitenMatha Amrithananda Mayisreeraj.guruvayoorNoch keine Bewertungen
- VillageExtensionofficer PMDDokument6 SeitenVillageExtensionofficer PMDPrasanth PnairNoch keine Bewertungen
- Five Secrets of SuccessDokument24 SeitenFive Secrets of Successsreeraj.guruvayoorNoch keine Bewertungen
- DKarmaDokument8 SeitenDKarmasreeraj.guruvayoorNoch keine Bewertungen
- Coordination Chemistry IIDokument77 SeitenCoordination Chemistry IIMaheshNoch keine Bewertungen
- SirshasanaaDokument4 SeitenSirshasanaasreeraj.guruvayoorNoch keine Bewertungen
- Get SuccessDokument13 SeitenGet Successsreeraj.guruvayoorNoch keine Bewertungen
- Engineering ResumesDokument13 SeitenEngineering Resumessreeraj.guruvayoorNoch keine Bewertungen
- Furnising of Information in Respect of Financial Support 2013 PDFDokument1 SeiteFurnising of Information in Respect of Financial Support 2013 PDFsreeraj.guruvayoorNoch keine Bewertungen
- Braham SamadhiDokument2 SeitenBraham Samadhisreeraj.guruvayoorNoch keine Bewertungen
- Luke 6 38Dokument8 SeitenLuke 6 38sreeraj.guruvayoorNoch keine Bewertungen
- Swami VivekanandaDokument10 SeitenSwami Vivekanandaprateek43535Noch keine Bewertungen
- Checklist Documenting Informed Consent 1336679958Dokument1 SeiteChecklist Documenting Informed Consent 1336679958sreeraj.guruvayoorNoch keine Bewertungen
- SOPDokument253 SeitenSOPMuhammad Younis BhatNoch keine Bewertungen
- The Drug Approval ProcessDokument7 SeitenThe Drug Approval Processsreeraj.guruvayoorNoch keine Bewertungen
- 1 1 Production Review and Approval of Jcto SopsDokument6 Seiten1 1 Production Review and Approval of Jcto SopsSreeraj Guruvayoor SNoch keine Bewertungen
- Site FDA inspection checklistDokument9 SeitenSite FDA inspection checklistsreeraj.guruvayoorNoch keine Bewertungen
- UoA-NHSG-SOP-014 - V2.00 - Recording Managing and Reporting AEs SAEs and SUSARsDokument14 SeitenUoA-NHSG-SOP-014 - V2.00 - Recording Managing and Reporting AEs SAEs and SUSARssreeraj.guruvayoorNoch keine Bewertungen
- 6 PreliminariesDokument21 Seiten6 PreliminariesJohn KellyNoch keine Bewertungen
- SOP 2-2 Obtaining Informed Consent FinalDokument7 SeitenSOP 2-2 Obtaining Informed Consent Finalsreeraj.guruvayoorNoch keine Bewertungen
- Anatomy GCP Insp TothAllen FDA USADokument19 SeitenAnatomy GCP Insp TothAllen FDA USAsreeraj.guruvayoorNoch keine Bewertungen
- CRA S Guide To MonitoringDokument224 SeitenCRA S Guide To Monitoringsreeraj.guruvayoor100% (6)
- Patient Selection in Clinical Trials PDFDokument90 SeitenPatient Selection in Clinical Trials PDFsreeraj.guruvayoorNoch keine Bewertungen
- Narrative WrittingDokument90 SeitenNarrative Writtingsreeraj.guruvayoorNoch keine Bewertungen
- Sample GCP ChecklistDokument8 SeitenSample GCP Checklistsreeraj.guruvayoor100% (1)
- Give It Will Be Given To YouDokument14 SeitenGive It Will Be Given To Yousreeraj.guruvayoorNoch keine Bewertungen
- A Practical Guide On Pharmacovigilance For BeginnersDokument10 SeitenA Practical Guide On Pharmacovigilance For Beginnerssreeraj.guruvayoorNoch keine Bewertungen
- Quality of VigiBase Reports Vital for Patient SafetyDokument0 SeitenQuality of VigiBase Reports Vital for Patient Safetysreeraj.guruvayoorNoch keine Bewertungen
- Ivrs Interactive Voice Response SystemDokument3 SeitenIvrs Interactive Voice Response Systemsreeraj.guruvayoorNoch keine Bewertungen
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeVon EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeBewertung: 4 von 5 Sternen4/5 (5782)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceVon EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceBewertung: 4 von 5 Sternen4/5 (890)
- The Yellow House: A Memoir (2019 National Book Award Winner)Von EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Bewertung: 4 von 5 Sternen4/5 (98)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureVon EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureBewertung: 4.5 von 5 Sternen4.5/5 (474)
- Shoe Dog: A Memoir by the Creator of NikeVon EverandShoe Dog: A Memoir by the Creator of NikeBewertung: 4.5 von 5 Sternen4.5/5 (537)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaVon EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaBewertung: 4.5 von 5 Sternen4.5/5 (265)
- The Little Book of Hygge: Danish Secrets to Happy LivingVon EverandThe Little Book of Hygge: Danish Secrets to Happy LivingBewertung: 3.5 von 5 Sternen3.5/5 (399)
- Never Split the Difference: Negotiating As If Your Life Depended On ItVon EverandNever Split the Difference: Negotiating As If Your Life Depended On ItBewertung: 4.5 von 5 Sternen4.5/5 (838)
- Grit: The Power of Passion and PerseveranceVon EverandGrit: The Power of Passion and PerseveranceBewertung: 4 von 5 Sternen4/5 (587)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryVon EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryBewertung: 3.5 von 5 Sternen3.5/5 (231)
- The Emperor of All Maladies: A Biography of CancerVon EverandThe Emperor of All Maladies: A Biography of CancerBewertung: 4.5 von 5 Sternen4.5/5 (271)
- Team of Rivals: The Political Genius of Abraham LincolnVon EverandTeam of Rivals: The Political Genius of Abraham LincolnBewertung: 4.5 von 5 Sternen4.5/5 (234)
- On Fire: The (Burning) Case for a Green New DealVon EverandOn Fire: The (Burning) Case for a Green New DealBewertung: 4 von 5 Sternen4/5 (72)
- The Unwinding: An Inner History of the New AmericaVon EverandThe Unwinding: An Inner History of the New AmericaBewertung: 4 von 5 Sternen4/5 (45)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersVon EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersBewertung: 4.5 von 5 Sternen4.5/5 (344)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyVon EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyBewertung: 3.5 von 5 Sternen3.5/5 (2219)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreVon EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreBewertung: 4 von 5 Sternen4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Von EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Bewertung: 4.5 von 5 Sternen4.5/5 (119)
- Her Body and Other Parties: StoriesVon EverandHer Body and Other Parties: StoriesBewertung: 4 von 5 Sternen4/5 (821)
- CMBS World - Reremic PhenomenonDokument14 SeitenCMBS World - Reremic PhenomenonykkwonNoch keine Bewertungen
- Fundamental of ICT: Study PresentationDokument12 SeitenFundamental of ICT: Study PresentationBikal ShresthaNoch keine Bewertungen
- AWS ServicesDokument25 SeitenAWS ServicesShaik Mahammad AlthafNoch keine Bewertungen
- A Handbook for Cooperative Fieldworkers in Developing NationsDokument389 SeitenA Handbook for Cooperative Fieldworkers in Developing NationsjimborenoNoch keine Bewertungen
- 2018 Bar Examinations Practical Exercises: Legal and Judicial Ethics andDokument9 Seiten2018 Bar Examinations Practical Exercises: Legal and Judicial Ethics andrfylananNoch keine Bewertungen
- The Marcos Ill-Gotten WealthDokument12 SeitenThe Marcos Ill-Gotten WealthAngel Nieto PengsonNoch keine Bewertungen
- ECC Chemical Process Pumps GuideDokument84 SeitenECC Chemical Process Pumps GuideIwan KurniawanNoch keine Bewertungen
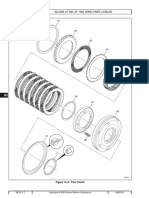
- Allison at 500, at 1500 Series Parts CatalogDokument3 SeitenAllison at 500, at 1500 Series Parts Catalogamin chaabenNoch keine Bewertungen
- Eco System and Green Logistics BasicsDokument32 SeitenEco System and Green Logistics BasicsBudmed GanbaatarNoch keine Bewertungen
- Division 8 Doors and Windows: Group 3Dokument22 SeitenDivision 8 Doors and Windows: Group 3Alen Ybanez100% (2)
- Skse ReadmeDokument3 SeitenSkse ReadmeJayseDVSNoch keine Bewertungen
- Retail Design: Prepared by Dilbar IqbalDokument16 SeitenRetail Design: Prepared by Dilbar IqbalDILBAR SHAKIRNoch keine Bewertungen
- Comparison of ROBOT and MIDAS GENDokument2 SeitenComparison of ROBOT and MIDAS GENAshish LoyaNoch keine Bewertungen
- Bangalisan V Ca G.R. No. 124678 July 31, 1997 Regalado, J.: FactsDokument1 SeiteBangalisan V Ca G.R. No. 124678 July 31, 1997 Regalado, J.: FactsKate GaroNoch keine Bewertungen
- The Risk Management of Medical Device-Related Pressure Ulcers Based On The Australian/ New Zealand StandardDokument11 SeitenThe Risk Management of Medical Device-Related Pressure Ulcers Based On The Australian/ New Zealand StandardJugurtha BoutlikhetNoch keine Bewertungen
- Learjet 60 MMEL Revision 5 SummaryDokument102 SeitenLearjet 60 MMEL Revision 5 SummaryHonorio Perez MNoch keine Bewertungen
- Fri Chiks Marketing Mix and Swot AnalysisDokument22 SeitenFri Chiks Marketing Mix and Swot Analysisahsanparveen100% (1)
- Case of Harvey V FaceyDokument2 SeitenCase of Harvey V Faceyveera89% (18)
- B1 Editable Quiz 9Dokument2 SeitenB1 Editable Quiz 9Dzima Simona100% (1)
- DP Event Bulletin 04 23Dokument14 SeitenDP Event Bulletin 04 23Sorin VirlanNoch keine Bewertungen
- Micropipettes SOPDokument1 SeiteMicropipettes SOPNishchaya SinghNoch keine Bewertungen
- Introduction To Distributed SystemsDokument5 SeitenIntroduction To Distributed SystemsoljiraaNoch keine Bewertungen
- Five Core Competencies of The Lean EnterpriseDokument3 SeitenFive Core Competencies of The Lean EnterpriseSai Sunil ChandraaNoch keine Bewertungen
- HP MOQ Traditional BPC Mar'21 Pricelist - FTPDokument8 SeitenHP MOQ Traditional BPC Mar'21 Pricelist - FTPrachamreddyrNoch keine Bewertungen
- Quadcopter Project Presentation Using KK 2.1.5 ControllerDokument13 SeitenQuadcopter Project Presentation Using KK 2.1.5 ControllerAkshay SinghalNoch keine Bewertungen
- COMLAWREV Catindig 2009 ReviewerDokument196 SeitenCOMLAWREV Catindig 2009 ReviewerShara LynNoch keine Bewertungen
- SIT 6404 57th Ave Report - Final052913Dokument301 SeitenSIT 6404 57th Ave Report - Final052913epraetorianNoch keine Bewertungen
- Profile of FAOADokument5 SeitenProfile of FAOAqubrex1Noch keine Bewertungen
- User Manual FADALDokument583 SeitenUser Manual FADALAntonio GonçalvesNoch keine Bewertungen
- Utah County Attorney Prioritized CrimesDokument9 SeitenUtah County Attorney Prioritized CrimesThe Salt Lake TribuneNoch keine Bewertungen