Das könnte Ihnen auch gefallen
- Tutorial Docking With GlideDokument5 SeitenTutorial Docking With GlideJohn M. E. PurbaNoch keine Bewertungen
- Essential Algorithms: A Practical Approach to Computer AlgorithmsVon EverandEssential Algorithms: A Practical Approach to Computer AlgorithmsBewertung: 4.5 von 5 Sternen4.5/5 (2)
- Franc3D V6 Ansys Fretting TutorialDokument15 SeitenFranc3D V6 Ansys Fretting Tutorialronald_edinsonNoch keine Bewertungen
- Molecular Modeling of Proteins PDFDokument474 SeitenMolecular Modeling of Proteins PDFFlorAyquiCueva100% (1)
- Dot Point IB Biology - AHL - Kerri Humphreys - Science 2010 PDFDokument50 SeitenDot Point IB Biology - AHL - Kerri Humphreys - Science 2010 PDFJulio Cèsar Torres Hernández100% (1)
- 7 Analysis of A Composite Aircraft Structure Using PCOMPGDokument9 Seiten7 Analysis of A Composite Aircraft Structure Using PCOMPGVinoth BalasubramaniyanNoch keine Bewertungen
- Synthetic Protein Switches Methods and Protocols (Methods in Molecular Biology)Dokument332 SeitenSynthetic Protein Switches Methods and Protocols (Methods in Molecular Biology)phyaravi100% (1)
- Nuclear Magnetic Resonance Spectros PDFDokument310 SeitenNuclear Magnetic Resonance Spectros PDFValemtinoNoch keine Bewertungen
- Gromacs Tutorial For Drug Enzyme ComplexDokument21 SeitenGromacs Tutorial For Drug Enzyme ComplexManoj Kumar RoutNoch keine Bewertungen
- Molecular Docking Using AutoDock and MGL Tools - GirinathDokument23 SeitenMolecular Docking Using AutoDock and MGL Tools - GirinathGirinath Pillai60% (5)
- Phast - Adding A Component From DIPPR PDFDokument22 SeitenPhast - Adding A Component From DIPPR PDFelvithaf100% (1)
- Aspen IQ ModelDokument71 SeitenAspen IQ Modelhaseeb100% (1)
- DOCK6.1 TutorialDokument15 SeitenDOCK6.1 Tutorial@lsreshtyNoch keine Bewertungen
- 105 Machine Learning PaperDokument6 Seiten105 Machine Learning PaperAabda AhmedNoch keine Bewertungen
- 3 Protein Structure-S PDFDokument10 Seiten3 Protein Structure-S PDFNathan Pittman0% (1)
- Molecular Biology of The Cell, 5th EditionDokument82 SeitenMolecular Biology of The Cell, 5th EditionBee Nunes24% (66)
- Asp .Net MVCDokument114 SeitenAsp .Net MVCFernando Torrez100% (3)
- Glucose OxidaseDokument42 SeitenGlucose OxidaseShauqah Mawaddah0% (1)
- Application of Data Mining in BioinformaticsDokument5 SeitenApplication of Data Mining in BioinformaticsYuni ListianaNoch keine Bewertungen
- Fundamentals of Enzymology The Cell and Molecular Biology of Catalytic ProteinsDokument0 SeitenFundamentals of Enzymology The Cell and Molecular Biology of Catalytic Proteinsmonica_elizabeth_35Noch keine Bewertungen
- DockingDokument6 SeitenDockingFrancimauro MoraisNoch keine Bewertungen
- Auto Dock TutorialDokument2 SeitenAuto Dock TutorialParijat DasNoch keine Bewertungen
- Re Cross Docking Tutorial 27aug12Dokument5 SeitenRe Cross Docking Tutorial 27aug12dulNoch keine Bewertungen
- Autodock Tutorial Paso A PasoDokument32 SeitenAutodock Tutorial Paso A PasoFabio CastellanosNoch keine Bewertungen
- ) AutoLigand TutorialDokument26 Seiten) AutoLigand TutorialClaudio Alejandro PereiraNoch keine Bewertungen
- Argus Molecular Docking 1Dokument5 SeitenArgus Molecular Docking 1escherichioNoch keine Bewertungen
- AutoDock TutorialDokument3 SeitenAutoDock Tutorialapi-382392950% (2)
- Protein structure modification using SPDBVDokument10 SeitenProtein structure modification using SPDBVPrayukta PadelkarNoch keine Bewertungen
- 11 Molecular DockingDokument6 Seiten11 Molecular DockingCarissa IslaNoch keine Bewertungen
- Basic Tutorial DockThor 1.0 6Dokument17 SeitenBasic Tutorial DockThor 1.0 6Emmanuel MarinhoNoch keine Bewertungen
- Modul DockingDokument14 SeitenModul DockingSagung DyahNoch keine Bewertungen
- Using Autodock With Autodocktools: A Tutorial: Written by Ruth Huey and Garrett M. MorrisDokument69 SeitenUsing Autodock With Autodocktools: A Tutorial: Written by Ruth Huey and Garrett M. MorrisIsa AlojNoch keine Bewertungen
- Manual BioblenderDokument28 SeitenManual BioblenderFernando Henrique DeslockNoch keine Bewertungen
- Autodock - Vina ProtocolDokument12 SeitenAutodock - Vina ProtocolNaresh kumarNoch keine Bewertungen
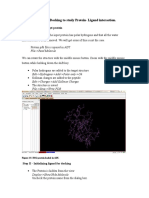
- Molecular Docking Tostudy Protein-Ligand InteractionDokument5 SeitenMolecular Docking Tostudy Protein-Ligand InteractionDr. Kaushal Kishor SharmaNoch keine Bewertungen
- 2012 DOCK Tutorial With StreptavidinDokument18 Seiten2012 DOCK Tutorial With Streptavidinboy20_bh100% (1)
- Protein Structure ExplorationDokument22 SeitenProtein Structure Explorationsuveer698Noch keine Bewertungen
- Guidelines and QuestionsDokument6 SeitenGuidelines and Questionsd nNoch keine Bewertungen
- Petrolog ManualDokument23 SeitenPetrolog ManualNibin TomNoch keine Bewertungen
- Institutional Summer Training WorksheetDokument9 SeitenInstitutional Summer Training WorksheetApoorva SinghNoch keine Bewertungen
- Docking TutorialDokument48 SeitenDocking Tutorialapi-3823929100% (4)
- Altera Max Plus TutorialDokument15 SeitenAltera Max Plus TutorialMarwan AhmedNoch keine Bewertungen
- Illustrated Step-By-Step Protocol To Perform Molecular DockingDokument64 SeitenIllustrated Step-By-Step Protocol To Perform Molecular DockingMasyitah ulfayuraNoch keine Bewertungen
- 5700 MlogitDokument38 Seiten5700 MlogitNawaz AliNoch keine Bewertungen
- PyMol PracticalDokument7 SeitenPyMol PracticalTom FlemingNoch keine Bewertungen
- Chimera ProceduresDokument3 SeitenChimera Proceduresraj_shiv10Noch keine Bewertungen
- OS-T - 2040 Spot Weld PDFDokument4 SeitenOS-T - 2040 Spot Weld PDFHrishikesh PhadkeNoch keine Bewertungen
- Modal Transient Dynamic Analysis of A BracketDokument8 SeitenModal Transient Dynamic Analysis of A BracketSrikanth Kabbal MNoch keine Bewertungen
- Arguslab NavodilaDokument6 SeitenArguslab NavodilaChandrakanthNoch keine Bewertungen
- AutoDock Tutorial v1.2Dokument3 SeitenAutoDock Tutorial v1.2Judith NelsonNoch keine Bewertungen
- Using Modal Analysis: ExerciseDokument5 SeitenUsing Modal Analysis: ExercisePraveen SreedharanNoch keine Bewertungen
- Replay A Log On Petri Net For Conformance Analysis Plug-In PDFDokument11 SeitenReplay A Log On Petri Net For Conformance Analysis Plug-In PDFNovandi BanitamaNoch keine Bewertungen
- DSP LAB - IntroductionDokument29 SeitenDSP LAB - IntroductionrkNoch keine Bewertungen
- Dock and Score Tyrosine Kinase InhibitorsDokument14 SeitenDock and Score Tyrosine Kinase InhibitorsvarinderkumarscribdNoch keine Bewertungen
- PyRx Manual PDFDokument13 SeitenPyRx Manual PDFநமச்சிவாயம் பாலகிருஷ்ணன்100% (2)
- Autodock ProtocolDokument17 SeitenAutodock ProtocolSasikala RajendranNoch keine Bewertungen
- Fatigue Analysis (Damage Calculation) Using S-N ApproachDokument7 SeitenFatigue Analysis (Damage Calculation) Using S-N ApproachdhanrajNoch keine Bewertungen
- ADT Autodock TutorialDokument51 SeitenADT Autodock TutorialelmozzNoch keine Bewertungen
- 01 - Lab - 1- Mô phỏng ADSDokument19 Seiten01 - Lab - 1- Mô phỏng ADSNguyễn Ngọc TháiNoch keine Bewertungen
- Input/Edit Core DipsDokument12 SeitenInput/Edit Core DipsKopet GaringNoch keine Bewertungen
- Introduction To Codewarrior™ - Simulating The Microcontroller in Assembly LanguageDokument9 SeitenIntroduction To Codewarrior™ - Simulating The Microcontroller in Assembly LanguageChristian CruzNoch keine Bewertungen
- ChemSep Tutorial: Understanding PCDmanagerDokument37 SeitenChemSep Tutorial: Understanding PCDmanagerjoseNoch keine Bewertungen
- WinHYDROPRO 10.54 user guideDokument4 SeitenWinHYDROPRO 10.54 user guideferdinanfirdausNoch keine Bewertungen
- Getting Started With Postgis: TopicDokument30 SeitenGetting Started With Postgis: TopicSAMINA ATTARINoch keine Bewertungen
- PyrxDokument13 SeitenPyrxahsaanahmadNoch keine Bewertungen
- C++ RefcardDokument2 SeitenC++ RefcardmaxNoch keine Bewertungen
- C++ Short NoteDokument6 SeitenC++ Short Notejohn07bbbNoch keine Bewertungen
- Programming in C Sharp Course Great GuidDokument3 SeitenProgramming in C Sharp Course Great GuidrednriNoch keine Bewertungen
- Power ShellDokument3 SeitenPower ShellrednriNoch keine Bewertungen
- CNF Unit-IV Notes CSETUBE PDFDokument45 SeitenCNF Unit-IV Notes CSETUBE PDFrednriNoch keine Bewertungen
- Blg252e CompleteDokument561 SeitenBlg252e Completesuleman247Noch keine Bewertungen
- C++ Neural Networks and Fuzzy Logic - Valluru B. RaoDokument595 SeitenC++ Neural Networks and Fuzzy Logic - Valluru B. RaoThyago VasconcelosNoch keine Bewertungen
- C++ Notes on OOPDokument133 SeitenC++ Notes on OOPrednri0% (1)
- C Language Standard Data Types and OperatorsDokument2 SeitenC Language Standard Data Types and OperatorsPradeep KumarNoch keine Bewertungen
- Html5 TutorialDokument23 SeitenHtml5 TutorialHands OffNoch keine Bewertungen
- Cil2012 html5Dokument45 SeitenCil2012 html5sreejeshkeralamNoch keine Bewertungen
- CNF Unit-I Notes Csetube PDFDokument37 SeitenCNF Unit-I Notes Csetube PDFthebhas1954Noch keine Bewertungen
- What You'll Build: by Scott Hanselman - January 12, 2011Dokument334 SeitenWhat You'll Build: by Scott Hanselman - January 12, 2011Ebenezer Tweneboah-KoduahNoch keine Bewertungen
- Adafruit Pitft 3 Dot 5 Touch Screen For Raspberry Pi PDFDokument50 SeitenAdafruit Pitft 3 Dot 5 Touch Screen For Raspberry Pi PDFrednri100% (1)
- 2609a Delivery Guide PDFDokument748 Seiten2609a Delivery Guide PDFrednriNoch keine Bewertungen
- AJAX Weather, JQuery Effects, AngularJS City AppDokument4 SeitenAJAX Weather, JQuery Effects, AngularJS City ApprednriNoch keine Bewertungen
- ExercisesDokument3 SeitenExercisesrednriNoch keine Bewertungen
- 15 Introduction of Softcomputing Approach in Slope StabilityDokument8 Seiten15 Introduction of Softcomputing Approach in Slope StabilityrednriNoch keine Bewertungen
- 09 - Chapter 3 PDFDokument80 Seiten09 - Chapter 3 PDFrednriNoch keine Bewertungen
- ASP NET 4 and Visual Studio 2010 Web Development OverviewDokument66 SeitenASP NET 4 and Visual Studio 2010 Web Development OverviewKalai VananNoch keine Bewertungen
- ExercisesDokument3 SeitenExercisesrednriNoch keine Bewertungen
- Developing Algorithmic and Logical Thinking SkillsDokument11 SeitenDeveloping Algorithmic and Logical Thinking SkillsrednriNoch keine Bewertungen
- The STRCHR Function: Using STRCHR To Search A String For A Single CharacterDokument20 SeitenThe STRCHR Function: Using STRCHR To Search A String For A Single CharacterrednriNoch keine Bewertungen
- Hidden Markov Models in Bioinformatics: Example Domain: Gene FindingDokument32 SeitenHidden Markov Models in Bioinformatics: Example Domain: Gene FindingrednriNoch keine Bewertungen
- Introduction To S AsDokument16 SeitenIntroduction To S AsrednriNoch keine Bewertungen
- SAS Guide Revf10Dokument12 SeitenSAS Guide Revf10rednriNoch keine Bewertungen
- A Brief Introduction To SAS Operators and FunctionsDokument7 SeitenA Brief Introduction To SAS Operators and FunctionsrednriNoch keine Bewertungen
- Biomolecules and Its UsesDokument20 SeitenBiomolecules and Its UsesRajendra Swarnakar25% (4)
- In Silico Study of Single Chain Fragment Variable (SCFV) On Chikungunya Virus Using Indonesian StrainDokument5 SeitenIn Silico Study of Single Chain Fragment Variable (SCFV) On Chikungunya Virus Using Indonesian StrainAdriani HasyimNoch keine Bewertungen
- Full Download Test Bank For Molecular Cell Biology Eighth Edition PDF Full ChapterDokument36 SeitenFull Download Test Bank For Molecular Cell Biology Eighth Edition PDF Full Chapterscottthorntongjpnfeatrw100% (18)
- A Simple Method For Displaying The Hydropathic Character of A ProteinDokument28 SeitenA Simple Method For Displaying The Hydropathic Character of A ProteinBiosynthesisNoch keine Bewertungen
- Proteins: Their Biological Functions and Primary StructureDokument59 SeitenProteins: Their Biological Functions and Primary StructureIsha BhartiNoch keine Bewertungen
- Structural Changes in Cellulose During PDokument8 SeitenStructural Changes in Cellulose During ParjunanpnNoch keine Bewertungen
- Biology HL Past Papers GuideDokument90 SeitenBiology HL Past Papers GuideelenaNoch keine Bewertungen
- Biochemistry Check for Understanding QuestionsDokument6 SeitenBiochemistry Check for Understanding QuestionsG INoch keine Bewertungen
- CV Alberto PerezDokument5 SeitenCV Alberto Perezaenon99Noch keine Bewertungen
- Chapter 4 Protein Three-Dimensional StructureDokument24 SeitenChapter 4 Protein Three-Dimensional StructureRO OMNoch keine Bewertungen
- 2001 Dobson Gerrard Pratt Foundations of Chemical BiologyDokument110 Seiten2001 Dobson Gerrard Pratt Foundations of Chemical BiologyElmer L'éléphantNoch keine Bewertungen
- Vanders Human Physiology The Mechanisms of Body Function 13th Edition Widmaier Solutions ManualDokument26 SeitenVanders Human Physiology The Mechanisms of Body Function 13th Edition Widmaier Solutions ManualJadeFischerqtcj100% (29)
- UniTartuCS Poster Template PortraitDokument1 SeiteUniTartuCS Poster Template PortraitManuel ZumbadoNoch keine Bewertungen
- Lenoir FlowProcessFold PDFDokument29 SeitenLenoir FlowProcessFold PDFMohammed Gism AllahNoch keine Bewertungen
- A Review On New Target Insulysine (Ide) & Its Inhibitors As Antidiabetics AgentsDokument12 SeitenA Review On New Target Insulysine (Ide) & Its Inhibitors As Antidiabetics AgentsTJPRC PublicationsNoch keine Bewertungen
- Halder2020 hydrophilicAminoAcidsProteinEthanolDokument10 SeitenHalder2020 hydrophilicAminoAcidsProteinEthanolSBNoch keine Bewertungen
- Accurate Calculation of Free Energy Changes Upon Amino Acid MutationDokument29 SeitenAccurate Calculation of Free Energy Changes Upon Amino Acid MutationThales FreireNoch keine Bewertungen
- CHEMICALS OF LIFE @SCZ Salongo 2016 PDFDokument18 SeitenCHEMICALS OF LIFE @SCZ Salongo 2016 PDFKlint Rodney Mark LuleNoch keine Bewertungen
- Proteins: Ms. Jirehkriza G. Suganob General Biology 1Dokument41 SeitenProteins: Ms. Jirehkriza G. Suganob General Biology 1Jcob SntosNoch keine Bewertungen
- Biotechnology SyllabusDokument91 SeitenBiotechnology Syllabuskiran.rNoch keine Bewertungen
- Protein StructureDokument11 SeitenProtein Structurelol heyNoch keine Bewertungen
- Aquasome: A Novel Drug DeliveryDokument11 SeitenAquasome: A Novel Drug DeliveryIjupbs IjupbsNoch keine Bewertungen