Das könnte Ihnen auch gefallen
- US 0017321 DuPontLammot GunpowderImprovement 19may1857Dokument1 SeiteUS 0017321 DuPontLammot GunpowderImprovement 19may1857jazmurdochNoch keine Bewertungen
- US Patent For Retractable Dog Leash From 1908Dokument3 SeitenUS Patent For Retractable Dog Leash From 1908jazmurdochNoch keine Bewertungen
- EP 0438426B1 MurdochJosephR PolyLactideCompositions 18nov1993Dokument19 SeitenEP 0438426B1 MurdochJosephR PolyLactideCompositions 18nov1993jazmurdochNoch keine Bewertungen
- EP 0438426B1 MurdochJosephR PolyLactideCompositions 18nov1993Dokument19 SeitenEP 0438426B1 MurdochJosephR PolyLactideCompositions 18nov1993jazmurdochNoch keine Bewertungen
- The Low Ionization Potentials of Guanine by Photoelectron SpectrosDokument6 SeitenThe Low Ionization Potentials of Guanine by Photoelectron SpectrosjazmurdochNoch keine Bewertungen
- MurdochJR BidentateLigands JACS 1984 106 4717Dokument7 SeitenMurdochJR BidentateLigands JACS 1984 106 4717jazmurdochNoch keine Bewertungen
- MurdochJR DLiBN JACS 1984 106 7843Dokument3 SeitenMurdochJR DLiBN JACS 1984 106 7843jazmurdochNoch keine Bewertungen
- US 4766182 PolylactideCompositions 23aug1988Dokument10 SeitenUS 4766182 PolylactideCompositions 23aug1988jazmurdochNoch keine Bewertungen
- LE Flory Patents Electronic Computing Device in 1946Dokument9 SeitenLE Flory Patents Electronic Computing Device in 1946jazmurdochNoch keine Bewertungen
- Robert Goddard Secures Rocket Patent in 1914Dokument4 SeitenRobert Goddard Secures Rocket Patent in 1914jazmurdochNoch keine Bewertungen
- Multi-OpticalDetectionSystem US 5268305 PDFDokument13 SeitenMulti-OpticalDetectionSystem US 5268305 PDFjazmurdochNoch keine Bewertungen
- ZwickyF OnThePossibleInfluenceOfTheMosaicStructureOfCrystalsOnAvogadro'sNumber PNAS 1930 16-3-211-5Dokument5 SeitenZwickyF OnThePossibleInfluenceOfTheMosaicStructureOfCrystalsOnAvogadro'sNumber PNAS 1930 16-3-211-5jazmurdochNoch keine Bewertungen
- MurdochJR RelationshipBetwEnergyAdd&EquivGroup JACS 1982 104 2782Dokument8 SeitenMurdochJR RelationshipBetwEnergyAdd&EquivGroup JACS 1982 104 2782jazmurdochNoch keine Bewertungen
- What Is The Rate-Limiting Step A Multistep Reaction?: With StableDokument5 SeitenWhat Is The Rate-Limiting Step A Multistep Reaction?: With StablejazmurdochNoch keine Bewertungen
- MurdochJR Hemistruct JACS 1982 104 588Dokument13 SeitenMurdochJR Hemistruct JACS 1982 104 588jazmurdochNoch keine Bewertungen
- Tau - Property Summary - s035Dokument80 SeitenTau - Property Summary - s035jazmurdochNoch keine Bewertungen
- KrivovSV FreeEnergySurface RevProteinFolding MDS PLoS 2009 5 7 E1000428Dokument10 SeitenKrivovSV FreeEnergySurface RevProteinFolding MDS PLoS 2009 5 7 E1000428jazmurdochNoch keine Bewertungen
- JonesC EndoEndo24Diphosphabicyclo110Butane Orbital Isomers CC 2001 663-4Dokument2 SeitenJonesC EndoEndo24Diphosphabicyclo110Butane Orbital Isomers CC 2001 663-4jazmurdochNoch keine Bewertungen
- BalakotaiahV DescrMicroMixing HomogeneousReactors ManuscriptDokument17 SeitenBalakotaiahV DescrMicroMixing HomogeneousReactors ManuscriptjazmurdochNoch keine Bewertungen
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceVon EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceBewertung: 4 von 5 Sternen4/5 (895)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeVon EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeBewertung: 4 von 5 Sternen4/5 (5794)
- The Yellow House: A Memoir (2019 National Book Award Winner)Von EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Bewertung: 4 von 5 Sternen4/5 (98)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureVon EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureBewertung: 4.5 von 5 Sternen4.5/5 (474)
- Shoe Dog: A Memoir by the Creator of NikeVon EverandShoe Dog: A Memoir by the Creator of NikeBewertung: 4.5 von 5 Sternen4.5/5 (537)
- The Little Book of Hygge: Danish Secrets to Happy LivingVon EverandThe Little Book of Hygge: Danish Secrets to Happy LivingBewertung: 3.5 von 5 Sternen3.5/5 (399)
- On Fire: The (Burning) Case for a Green New DealVon EverandOn Fire: The (Burning) Case for a Green New DealBewertung: 4 von 5 Sternen4/5 (73)
- Never Split the Difference: Negotiating As If Your Life Depended On ItVon EverandNever Split the Difference: Negotiating As If Your Life Depended On ItBewertung: 4.5 von 5 Sternen4.5/5 (838)
- Grit: The Power of Passion and PerseveranceVon EverandGrit: The Power of Passion and PerseveranceBewertung: 4 von 5 Sternen4/5 (588)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryVon EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryBewertung: 3.5 von 5 Sternen3.5/5 (231)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaVon EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaBewertung: 4.5 von 5 Sternen4.5/5 (266)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersVon EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersBewertung: 4.5 von 5 Sternen4.5/5 (344)
- The Emperor of All Maladies: A Biography of CancerVon EverandThe Emperor of All Maladies: A Biography of CancerBewertung: 4.5 von 5 Sternen4.5/5 (271)
- Team of Rivals: The Political Genius of Abraham LincolnVon EverandTeam of Rivals: The Political Genius of Abraham LincolnBewertung: 4.5 von 5 Sternen4.5/5 (234)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreVon EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreBewertung: 4 von 5 Sternen4/5 (1090)
- The Unwinding: An Inner History of the New AmericaVon EverandThe Unwinding: An Inner History of the New AmericaBewertung: 4 von 5 Sternen4/5 (45)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyVon EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyBewertung: 3.5 von 5 Sternen3.5/5 (2259)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Von EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Bewertung: 4.5 von 5 Sternen4.5/5 (120)
- Her Body and Other Parties: StoriesVon EverandHer Body and Other Parties: StoriesBewertung: 4 von 5 Sternen4/5 (821)
- Individual Reflection ScribdDokument4 SeitenIndividual Reflection ScribdJamie Chan JieminNoch keine Bewertungen
- 7273X 47 ITOW Mozart PDFDokument3 Seiten7273X 47 ITOW Mozart PDFAdrian KranjcevicNoch keine Bewertungen
- Job Sheet 1Dokument5 SeitenJob Sheet 1Sue AzizNoch keine Bewertungen
- Claudio MonteverdiDokument2 SeitenClaudio MonteverdiBrendan LynchNoch keine Bewertungen
- NUFLO Low Power Pre-Amplifier: SpecificationsDokument2 SeitenNUFLO Low Power Pre-Amplifier: SpecificationsJorge ParraNoch keine Bewertungen
- General Information Exhibition Guide Lines - 3P 2022Dokument6 SeitenGeneral Information Exhibition Guide Lines - 3P 2022muhammad khanNoch keine Bewertungen
- QLD Plan Draft Review Raw DataDokument242 SeitenQLD Plan Draft Review Raw DataRohit Jain100% (1)
- Statement of Cash Flows AnswerDokument3 SeitenStatement of Cash Flows Answeranber mohammadNoch keine Bewertungen
- Sample Spec For AWWA HDPE Pipe Fittings 6.02revDokument6 SeitenSample Spec For AWWA HDPE Pipe Fittings 6.02revmg4myNoch keine Bewertungen
- Daewoo SJ-210H DSJ-6000LHMDokument44 SeitenDaewoo SJ-210H DSJ-6000LHMMarco Antonio100% (5)
- MELASMA (Ardy, Kintan, Fransisca)Dokument20 SeitenMELASMA (Ardy, Kintan, Fransisca)Andi Firman MubarakNoch keine Bewertungen
- Sample Engagement LetterDokument5 SeitenSample Engagement Letterprincess_camarilloNoch keine Bewertungen
- SM-G900F Esquematico Completo Anibal Garcia IrepairDokument2 SeitenSM-G900F Esquematico Completo Anibal Garcia Irepairfix marketNoch keine Bewertungen
- Titus Selection of DiffuserDokument14 SeitenTitus Selection of DiffuserhanyassawyNoch keine Bewertungen
- Cyrille MATH INVESTIGATION Part2Dokument18 SeitenCyrille MATH INVESTIGATION Part2Jessie jorgeNoch keine Bewertungen
- Cinnamon RollDokument1 SeiteCinnamon RollMaria Manoa GantalaNoch keine Bewertungen
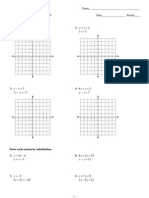
- Review Systems of Linear Equations All MethodsDokument4 SeitenReview Systems of Linear Equations All Methodsapi-265647260Noch keine Bewertungen
- Architech 06-2016 Room AssignmentDokument4 SeitenArchitech 06-2016 Room AssignmentPRC Baguio100% (1)
- Castle CrashesDokument21 SeitenCastle Crasheswicked wolfNoch keine Bewertungen
- Srinivasa Ramanujan - Britannica Online EncyclopediaDokument2 SeitenSrinivasa Ramanujan - Britannica Online EncyclopediaEvariste MigaboNoch keine Bewertungen
- SET UP Computer ServerDokument3 SeitenSET UP Computer ServerRicHArdNoch keine Bewertungen
- Quizlet Table 7Dokument1 SeiteQuizlet Table 7JosielynNoch keine Bewertungen
- R35 Credit Analysis Models - AnswersDokument13 SeitenR35 Credit Analysis Models - AnswersSakshiNoch keine Bewertungen
- Hilti TE 804 and 905avr PartsDokument13 SeitenHilti TE 804 and 905avr PartsAqui Solo100% (1)
- ISO 11064-12000 Ergonomic Design of Control Centres - Part 1 Principles For The Design of Control Centres by ISO TC 159SC 4WG 8Dokument6 SeitenISO 11064-12000 Ergonomic Design of Control Centres - Part 1 Principles For The Design of Control Centres by ISO TC 159SC 4WG 8marcianocalvi4611100% (2)
- Biography of Anna HazareDokument4 SeitenBiography of Anna HazareGenesis FirstNoch keine Bewertungen
- Thom22e ch03 FinalDokument44 SeitenThom22e ch03 FinalDionisius AlvianNoch keine Bewertungen
- No ApprovedDokument154 SeitenNo ApprovedAnnaNoch keine Bewertungen
- Module 1Dokument64 SeitenModule 1Jackyson RajkumarNoch keine Bewertungen
- 064 DIR - Launching Whipping Creme & Skimmed Milk Di Channel Horeka (Subdist Masuya)Dokument3 Seiten064 DIR - Launching Whipping Creme & Skimmed Milk Di Channel Horeka (Subdist Masuya)indra sapta PrahardikaNoch keine Bewertungen