Das könnte Ihnen auch gefallen
- Transition Metal Catalyzed Furans Synthesis: Transition Metal Catalyzed Heterocycle Synthesis SeriesVon EverandTransition Metal Catalyzed Furans Synthesis: Transition Metal Catalyzed Heterocycle Synthesis SeriesNoch keine Bewertungen
- Palladium-Catalyzed Hiyama Cross-Couplings of Aryl Arenesulfonates With ArylsilanesDokument2 SeitenPalladium-Catalyzed Hiyama Cross-Couplings of Aryl Arenesulfonates With ArylsilanessabafarooqNoch keine Bewertungen
- Angewandte: Gang Shan, Xinglin Yang, Linlin Ma, and Yu RaoDokument5 SeitenAngewandte: Gang Shan, Xinglin Yang, Linlin Ma, and Yu RaoBedanta BorahNoch keine Bewertungen
- Jacs AsapDokument2 SeitenJacs AsapMohamadMostafaviNoch keine Bewertungen
- Domino/knoevenagel-Hetero-Diels-Alder ReactionDokument12 SeitenDomino/knoevenagel-Hetero-Diels-Alder ReactionBhavesh PansuriyaNoch keine Bewertungen
- Importante-Anhidrido CHO Generan Multiheterociclo en CascadaDokument8 SeitenImportante-Anhidrido CHO Generan Multiheterociclo en CascadaFernando RSNoch keine Bewertungen
- Kinetics and Mechanisms of Homogeneous Catalytic Reactions. Part 12. Hydroalcoxycarbonylation of 1-Hexene Using Palladium/ Triphenylphosphine Systems As Catalyst PrecursorsDokument11 SeitenKinetics and Mechanisms of Homogeneous Catalytic Reactions. Part 12. Hydroalcoxycarbonylation of 1-Hexene Using Palladium/ Triphenylphosphine Systems As Catalyst PrecursorschelogkNoch keine Bewertungen
- Siegel Hydroxyl at I OnDokument5 SeitenSiegel Hydroxyl at I OncorechiNoch keine Bewertungen
- Eur. J, 2010, 16, 6509-6517 Reek Anti-HalpernDokument9 SeitenEur. J, 2010, 16, 6509-6517 Reek Anti-HalpernszbaloghNoch keine Bewertungen
- Highly e Cient Catalytic Dehydrative S-Allylation of Thiols and Thioic S-AcidswDokument3 SeitenHighly e Cient Catalytic Dehydrative S-Allylation of Thiols and Thioic S-AcidswPrasun Kanti PradhanNoch keine Bewertungen
- Pd/C-mediated synthesis of α-pyrone fused with a five-membered nitrogen heteroaryl ring: A new route to pyrano (4,3-c) pyrazol-4 (1H) -onesDokument5 SeitenPd/C-mediated synthesis of α-pyrone fused with a five-membered nitrogen heteroaryl ring: A new route to pyrano (4,3-c) pyrazol-4 (1H) -onesabdullahkhanduNoch keine Bewertungen
- MMC 1Dokument6 SeitenMMC 1umesh123patilNoch keine Bewertungen
- 1553 FTPDokument4 Seiten1553 FTPBedanta BorahNoch keine Bewertungen
- Full Paper: Min Han, Heng-Yi Zhang, Li-Xu Yang, Zhi-Jun Ding, Rui-Jie Zhuang, and Yu LiuDokument7 SeitenFull Paper: Min Han, Heng-Yi Zhang, Li-Xu Yang, Zhi-Jun Ding, Rui-Jie Zhuang, and Yu LiuKristine Dwi PuspitasariNoch keine Bewertungen
- Polyhedron: Dan-Ting Lu, Jiao He, Xiao-Yong Yu, Xu-Feng Liu, Yu-Long Li, Zhong-Qing JiangDokument6 SeitenPolyhedron: Dan-Ting Lu, Jiao He, Xiao-Yong Yu, Xu-Feng Liu, Yu-Long Li, Zhong-Qing JiangJackSchmeichelNoch keine Bewertungen
- Asian J. Org. Chem. 2021,10, 2152-2156Dokument5 SeitenAsian J. Org. Chem. 2021,10, 2152-2156NoimurNoch keine Bewertungen
- Synthesis of Anisole by Vapor Phase Methylation of Phenol With Methanol Over Catalysts Supported On Activated AluminaDokument3 SeitenSynthesis of Anisole by Vapor Phase Methylation of Phenol With Methanol Over Catalysts Supported On Activated AluminaNitish Singh SengarNoch keine Bewertungen
- Direct Synthesis of Methyl Formate From CO2 - 1 ALUMNDokument8 SeitenDirect Synthesis of Methyl Formate From CO2 - 1 ALUMNPamela Rivas LaraNoch keine Bewertungen
- Sdarticle 008Dokument29 SeitenSdarticle 008geo angNoch keine Bewertungen
- Synthesis of N - ( (Tert-Butoxy) Carbonyl) - 3 - (9,10-Dihydro-9-Oxoacridin-2-Yl) - L - Alanine, A Newfluorescent Amino Acid DerivativeDokument6 SeitenSynthesis of N - ( (Tert-Butoxy) Carbonyl) - 3 - (9,10-Dihydro-9-Oxoacridin-2-Yl) - L - Alanine, A Newfluorescent Amino Acid DerivativeVictor NgNoch keine Bewertungen
- Polystyrin Suported Al (OTF) 3Dokument3 SeitenPolystyrin Suported Al (OTF) 3shailesh19rajputNoch keine Bewertungen
- COM 08 11328asdfdsDokument9 SeitenCOM 08 11328asdfdsVictor NgNoch keine Bewertungen
- Poly EneDokument3 SeitenPoly EneMohammed TarekNoch keine Bewertungen
- Tetrahedron Letters: Olugbeminiyi O. Fadeyi, R. Nathan Daniels, Sean M. Deguire, Craig W. LindsleyDokument4 SeitenTetrahedron Letters: Olugbeminiyi O. Fadeyi, R. Nathan Daniels, Sean M. Deguire, Craig W. LindsleyNopasantiNoch keine Bewertungen
- Resume Jurnal Solid State ReactionDokument10 SeitenResume Jurnal Solid State ReactionSusantiNoch keine Bewertungen
- Highly Efficient One-pot Synthesis, Antimicrobial and Docking Studies of Newer β-amino Carbonyl Derivatives Catalyzed by Silica Sulfuric AcidDokument14 SeitenHighly Efficient One-pot Synthesis, Antimicrobial and Docking Studies of Newer β-amino Carbonyl Derivatives Catalyzed by Silica Sulfuric AcidnanoNoch keine Bewertungen
- Tetrahedron Letters 52 (2011) 5107-5109Dokument3 SeitenTetrahedron Letters 52 (2011) 5107-5109rajesh_tammana3550Noch keine Bewertungen
- Synthesis and Reactions of Some Heterocycles Containing Nitrogen and SulphurDokument15 SeitenSynthesis and Reactions of Some Heterocycles Containing Nitrogen and SulphuromansuNoch keine Bewertungen
- Synthetic CommunicationDokument7 SeitenSynthetic CommunicationDeepti AtluriNoch keine Bewertungen
- 549 553Dokument5 Seiten549 553Almaz KassNoch keine Bewertungen
- Chen 2015Dokument8 SeitenChen 2015Mohammad AhedNoch keine Bewertungen
- Kubicka Different SolventsDokument10 SeitenKubicka Different SolventscligcodiNoch keine Bewertungen
- Uses of 2-Ethoxy-4 (3H) Quinazolinone in Synthesis of Quinazoline and Quinazolinone Derivatives of Antimicrobial Activity: The Solvent EffectDokument12 SeitenUses of 2-Ethoxy-4 (3H) Quinazolinone in Synthesis of Quinazoline and Quinazolinone Derivatives of Antimicrobial Activity: The Solvent Effectkhaliddarwish1962Noch keine Bewertungen
- Novel Soluble and Thermally-Stable Fullerene Dyad Containing Perylene (Dokument5 SeitenNovel Soluble and Thermally-Stable Fullerene Dyad Containing Perylene (Lodrick WangatiaNoch keine Bewertungen
- Gian Luca Araldi Et Al - An Enantioselective Synthesis of 2-Alkyl-3-Phenyltropanes by An Asymmetric 1,3-Dipolar Cycloaddition ReactionDokument2 SeitenGian Luca Araldi Et Al - An Enantioselective Synthesis of 2-Alkyl-3-Phenyltropanes by An Asymmetric 1,3-Dipolar Cycloaddition ReactionGummyColaNoch keine Bewertungen
- Enantioselective Total Synthesis of (+) - Wortmannin: Yinliang Guo, Tianfei Quan, Yandong Lu, and Tuoping LuoDokument4 SeitenEnantioselective Total Synthesis of (+) - Wortmannin: Yinliang Guo, Tianfei Quan, Yandong Lu, and Tuoping LuoLê MinhNoch keine Bewertungen
- A Simple Approach Towards The Synthesis of OxadeazaflavinesDokument8 SeitenA Simple Approach Towards The Synthesis of OxadeazaflavinesThéoNoch keine Bewertungen
- Yamamoto Et Al. - 2009 - Γ-Selective Cross-Coupling Reactions of PotassiumDokument9 SeitenYamamoto Et Al. - 2009 - Γ-Selective Cross-Coupling Reactions of PotassiumVictor CiocalteaNoch keine Bewertungen
- Chem. Eur. J. 2011, 17, 10208 - 10212Dokument5 SeitenChem. Eur. J. 2011, 17, 10208 - 10212SBNoch keine Bewertungen
- Calcium Phosphate Apatites With Variable Ca/P Atomic Ratio I. Synthesis, Characterisation and Thermal Stability of PowdersDokument8 SeitenCalcium Phosphate Apatites With Variable Ca/P Atomic Ratio I. Synthesis, Characterisation and Thermal Stability of PowdersMaximimilianoNoch keine Bewertungen
- O Nucleophilic Amino Alcohol Acyl Transfer Catalysts The Effect of Acidity of The Hydroxyl Group On The Activity of The CatalystDokument4 SeitenO Nucleophilic Amino Alcohol Acyl Transfer Catalysts The Effect of Acidity of The Hydroxyl Group On The Activity of The Catalystbruenor304amancaboihNoch keine Bewertungen
- Cyclization-Activated Prodrugs. Synthesis, Reactivity and Toxicity of Dipeptide Esters of ParacetamolDokument4 SeitenCyclization-Activated Prodrugs. Synthesis, Reactivity and Toxicity of Dipeptide Esters of Paracetamollloi_25Noch keine Bewertungen
- One Electron Oxidation of 12-Tungstocobalt (III) Ate Microsomal Cytochrome P450Dokument4 SeitenOne Electron Oxidation of 12-Tungstocobalt (III) Ate Microsomal Cytochrome P450Narsinh M. DodiaNoch keine Bewertungen
- Comparison The Reactivity S - Adenylic Acid and S - Guanylic AcidDokument5 SeitenComparison The Reactivity S - Adenylic Acid and S - Guanylic AcidEr Mayur PatilNoch keine Bewertungen
- Bioorganic & Medicinal Chemistry Letters: Hanumantharao Paritala, Steven M. FirestineDokument4 SeitenBioorganic & Medicinal Chemistry Letters: Hanumantharao Paritala, Steven M. FirestineHadeel Al-SinjilawiNoch keine Bewertungen
- Book 3Dokument38 SeitenBook 3Amit ChakrabortyNoch keine Bewertungen
- A QMMM-Based Computational Investigation On The CatalyticMechanism of Saccharopine ReductaseDokument21 SeitenA QMMM-Based Computational Investigation On The CatalyticMechanism of Saccharopine ReductaseayeshaNoch keine Bewertungen
- Synthesis of Heteroaromatic Natural ProductsDokument127 SeitenSynthesis of Heteroaromatic Natural ProductsWilly CoioteNoch keine Bewertungen
- PNP Pourmousavi2017Dokument31 SeitenPNP Pourmousavi2017sonNoch keine Bewertungen
- Ring Opening Reaction of EpoxidesDokument5 SeitenRing Opening Reaction of Epoxides4t43t34yNoch keine Bewertungen
- Ocando Mavarez1998 PDFDokument7 SeitenOcando Mavarez1998 PDFMateus PinheiroNoch keine Bewertungen
- Chemical Engineering Journal Volume Issue 2019 (Doi 10.1016 - J.cej.2019.122237) Granado, Lérys Tavernier, Romain Foyer, Gabriel David, Ghisl - Catalysis For Highly Thermostable Phenol-TerephthalaDokument26 SeitenChemical Engineering Journal Volume Issue 2019 (Doi 10.1016 - J.cej.2019.122237) Granado, Lérys Tavernier, Romain Foyer, Gabriel David, Ghisl - Catalysis For Highly Thermostable Phenol-TerephthalasitiNoch keine Bewertungen
- Room Temperature C P Bond Formation Enabled by Merging Nickel Catalysis and Visible Light Induced Photoredox Catalysis - CompressDokument5 SeitenRoom Temperature C P Bond Formation Enabled by Merging Nickel Catalysis and Visible Light Induced Photoredox Catalysis - CompressshankhadeepawsNoch keine Bewertungen
- Dyes and Pigments-2016-New Class of Hyperpolarizable Push-Pull Organic Chromophores by Applying A Novel and Convenient Synthetic StrategyDokument5 SeitenDyes and Pigments-2016-New Class of Hyperpolarizable Push-Pull Organic Chromophores by Applying A Novel and Convenient Synthetic StrategyELKIN ALFONSO RODRIGUEZ AGUALIMPIANoch keine Bewertungen
- Catalytic Conversion of Propane To BTX Over Ga, ZN, Mo, Adn Re Impregnated ZSM-5 CatalystsDokument19 SeitenCatalytic Conversion of Propane To BTX Over Ga, ZN, Mo, Adn Re Impregnated ZSM-5 CatalystsJorge Gutierrez AranibarNoch keine Bewertungen
- NIH Public Access: Author ManuscriptDokument9 SeitenNIH Public Access: Author ManuscriptSyedFaridAliNoch keine Bewertungen
- Gas-Phase Hydroformylation of Propene Over Silica-Supported PPH - Modified Rhodium CatalystsDokument9 SeitenGas-Phase Hydroformylation of Propene Over Silica-Supported PPH - Modified Rhodium CatalystsIlireaNoch keine Bewertungen
- Li 2016Dokument9 SeitenLi 2016arash.abadianNoch keine Bewertungen
- (Elearnica - Ir) - Towards The Total Synthesis of Tashironin Related Allo-Cedrane Natural ProdDokument4 Seiten(Elearnica - Ir) - Towards The Total Synthesis of Tashironin Related Allo-Cedrane Natural ProdMohamadMostafaviNoch keine Bewertungen
- Cyclization-Activated Prodrugs. Synthesis, Reactivity and Toxicity of Dipeptide Esters of ParacetamolDokument0 SeitenCyclization-Activated Prodrugs. Synthesis, Reactivity and Toxicity of Dipeptide Esters of ParacetamolgsinamdarNoch keine Bewertungen
- Review de L-Prolina y Sus Derivados en Sintesis AsimetricaDokument31 SeitenReview de L-Prolina y Sus Derivados en Sintesis Asimetricatalero22Noch keine Bewertungen
- A 13C NMR and DSC Study of The Amorphous and Crystalline Phases in Asphalts (1999) - Parte Cristalina AsfaltosDokument9 SeitenA 13C NMR and DSC Study of The Amorphous and Crystalline Phases in Asphalts (1999) - Parte Cristalina Asfaltostalero22Noch keine Bewertungen
- Sintesis Asimetrica Con Aminas QuiralesDokument13 SeitenSintesis Asimetrica Con Aminas Quiralestalero22Noch keine Bewertungen
- Secondary Amine Synth ReviewDokument27 SeitenSecondary Amine Synth ReviewFacundo BaróNoch keine Bewertungen
- Prolina Catalizador de Reacciones AsimetricasDokument18 SeitenProlina Catalizador de Reacciones Asimetricastalero22Noch keine Bewertungen
- Synthesis of Mannish BasesDokument8 SeitenSynthesis of Mannish Basestalero22Noch keine Bewertungen
- Reacciones Tipo MannichDokument27 SeitenReacciones Tipo Mannichtalero22Noch keine Bewertungen
- WW-WASG03 Electrical Wire Sizes-WEB 7-7-11 PDFDokument1 SeiteWW-WASG03 Electrical Wire Sizes-WEB 7-7-11 PDFSemion VirtudazoNoch keine Bewertungen
- Completions and WorkoverDokument309 SeitenCompletions and WorkoverFan Jack67% (3)
- General Purpose ValvesDokument46 SeitenGeneral Purpose ValvesbataNoch keine Bewertungen
- Fyup Chemistry SyllabusDokument81 SeitenFyup Chemistry SyllabusRaj KumarNoch keine Bewertungen
- Ipa18 202 SeDokument15 SeitenIpa18 202 SeDimas Suryo WicaksonoNoch keine Bewertungen
- Fibc Type A B C D Classification SafetyDokument2 SeitenFibc Type A B C D Classification Safetydhineshbabu rNoch keine Bewertungen
- Unit 5.12 PrecipitationDokument16 SeitenUnit 5.12 PrecipitationMutale InongeNoch keine Bewertungen
- Adsc of Amorphous Sugar - Mettler ToledoDokument3 SeitenAdsc of Amorphous Sugar - Mettler ToledoMarthaLuceroPerezNoch keine Bewertungen
- Tisu Neural Neurofisiologi Neuron Neuroglia Terminologi SarafDokument141 SeitenTisu Neural Neurofisiologi Neuron Neuroglia Terminologi SarafRainne LeeNoch keine Bewertungen
- 12345Dokument1 Seite12345Praveen KumarNoch keine Bewertungen
- Assignment On: Textiles in Agriculture (Agrotech)Dokument6 SeitenAssignment On: Textiles in Agriculture (Agrotech)AmirParvezNoch keine Bewertungen
- Superalloys - A Primer and HistoryDokument4 SeitenSuperalloys - A Primer and Historyhemakumars100% (1)
- Wear Debris AnalysisDokument2 SeitenWear Debris Analysisthoma111sNoch keine Bewertungen
- Biomechanical Properties of A New Fiber-Reinforced CompositesDokument10 SeitenBiomechanical Properties of A New Fiber-Reinforced Compositesazam ahmedNoch keine Bewertungen
- Seta Verification Materials: STVM MTVMDokument2 SeitenSeta Verification Materials: STVM MTVMdchyNoch keine Bewertungen
- MK1977 CongressDokument173 SeitenMK1977 CongressGodshalllaughNoch keine Bewertungen
- 161Dokument7 Seiten161KierCliffenvilleGanadosPacienteNoch keine Bewertungen
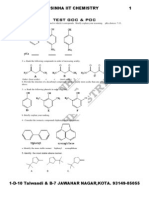
- Test1 Goc & Poc Tough by S.K.sinha See Chemistry Animations atDokument3 SeitenTest1 Goc & Poc Tough by S.K.sinha See Chemistry Animations atmyiitchemistry100% (1)
- Born Oppenheimer ApproximationDokument15 SeitenBorn Oppenheimer ApproximationElizabeth HarrisonNoch keine Bewertungen
- Presenters Post16 Tcm18-118246Dokument18 SeitenPresenters Post16 Tcm18-118246Kamariah IsmailNoch keine Bewertungen
- Hagglunds CaDokument19 SeitenHagglunds CaJonathan Giraldo100% (1)
- The Motion of ColorDokument6 SeitenThe Motion of Colorapi-374832521Noch keine Bewertungen
- Industrial ReportDokument64 SeitenIndustrial Reportfuad ullahNoch keine Bewertungen
- 5 Minute Guide Electricity StorageDokument17 Seiten5 Minute Guide Electricity StorageCarlos HolguinNoch keine Bewertungen
- Four Factors Affecting The Rate of Chemical ReactionDokument5 SeitenFour Factors Affecting The Rate of Chemical ReactionFeliciano Tristan E.Noch keine Bewertungen
- RingMethod Zuidema WatersDokument2 SeitenRingMethod Zuidema WatersJack Yoseph Martinez OrtegaNoch keine Bewertungen
- National Waste Management Strategy 2019-2023Dokument64 SeitenNational Waste Management Strategy 2019-2023Chikondi KanamaNoch keine Bewertungen
- Density MethodDokument5 SeitenDensity MethodMajed DawaNoch keine Bewertungen
- Sae 1025Dokument6 SeitenSae 1025Mada PerwiraNoch keine Bewertungen