Das könnte Ihnen auch gefallen
- Definition of BiopharmaceuticsDokument3 SeitenDefinition of BiopharmaceuticsvafaashkNoch keine Bewertungen
- Ch-3 Bioavailability of Drugs and Bioequivalence-18th BatchDokument30 SeitenCh-3 Bioavailability of Drugs and Bioequivalence-18th BatchFerdous Shahriar100% (1)
- Bioavailability and BioequivalenceDokument47 SeitenBioavailability and BioequivalenceGiovanne BuendiaNoch keine Bewertungen
- Pharmaceutical Equivalents: Nomenclature (As Excerpted From The Orange Book) Therapeutic Equivalence-Related TermsDokument4 SeitenPharmaceutical Equivalents: Nomenclature (As Excerpted From The Orange Book) Therapeutic Equivalence-Related TermskurutalaNoch keine Bewertungen
- Bioavailability and BioequivalenceDokument53 SeitenBioavailability and BioequivalenceWalaa abo foolNoch keine Bewertungen
- What Is Biopharmaceutics?: Brand NameDokument10 SeitenWhat Is Biopharmaceutics?: Brand NameAhmad Jamal HashmiNoch keine Bewertungen
- 8 Ba Be FdaDokument36 Seiten8 Ba Be Fdaraju narayana padalaNoch keine Bewertungen
- Final ProjectDokument119 SeitenFinal ProjectSaurabh KumarNoch keine Bewertungen
- Bioequivalence: Faculty of Pharmacy, Nursing and Health Professions Master in Industrial Pharmaceutical TechnologyDokument15 SeitenBioequivalence: Faculty of Pharmacy, Nursing and Health Professions Master in Industrial Pharmaceutical TechnologyMayson Bali100% (1)
- Ba-Be PDFDokument30 SeitenBa-Be PDFUswatun Hasanah7201Noch keine Bewertungen
- Bioavailability and BioequivalenceDokument56 SeitenBioavailability and Bioequivalenceنور الهدى100% (1)
- Guideline On Bioavailability and BioequivalanceDokument26 SeitenGuideline On Bioavailability and BioequivalanceGima Amezia SariNoch keine Bewertungen
- Bioequivalence: Faculty of Pharmacy, Nursing and Health Professions Master in Industrial Pharmaceutical TechnologyDokument16 SeitenBioequivalence: Faculty of Pharmacy, Nursing and Health Professions Master in Industrial Pharmaceutical TechnologyMayson BaliNoch keine Bewertungen
- Biopharmaceutics: Definitions and TerminologiesDokument30 SeitenBiopharmaceutics: Definitions and TerminologiesDanish KamalNoch keine Bewertungen
- IntroductionDokument71 SeitenIntroductionAreenub ArshadNoch keine Bewertungen
- Hemanagadurga2019 Bioavailability StudiesDokument17 SeitenHemanagadurga2019 Bioavailability Studiesraghuraj75Noch keine Bewertungen
- Bioequevalance StudiesDokument55 SeitenBioequevalance StudiesMubammad MursaleenNoch keine Bewertungen
- Formulary 2009Dokument51 SeitenFormulary 2009Wen ZhuNoch keine Bewertungen
- Review Article: Barbara Davit, April C. Braddy, Dale P. Conner, and Lawrence X. YuDokument17 SeitenReview Article: Barbara Davit, April C. Braddy, Dale P. Conner, and Lawrence X. Yulhthang1990Noch keine Bewertungen
- Non Linear PharmacokineticsDokument33 SeitenNon Linear PharmacokineticsClaudNoch keine Bewertungen
- Bioavailability and Bioequivalence (BABE)Dokument4 SeitenBioavailability and Bioequivalence (BABE)Wingielyn Honculada BaldozaNoch keine Bewertungen
- Biological Aspects of FormulationsDokument8 SeitenBiological Aspects of FormulationsKabirNoch keine Bewertungen
- Asean Guidelines For: Final Draft: 21 July 2004Dokument29 SeitenAsean Guidelines For: Final Draft: 21 July 2004Jone YingNoch keine Bewertungen
- Thailand BABE GuidelineDokument27 SeitenThailand BABE GuidelineDianeNoch keine Bewertungen
- Lecturenote - Bioavailability and BioequivalenceDokument40 SeitenLecturenote - Bioavailability and BioequivalenceAsnakeNoch keine Bewertungen
- 24 13v3i2 5Dokument9 Seiten24 13v3i2 5gopalNoch keine Bewertungen
- Difference Between Bioavailability and BioequivalenceDokument11 SeitenDifference Between Bioavailability and Bioequivalenceprakash deshmukhNoch keine Bewertungen
- ASEAN BE Guidelines-Amended According To1st BABE TWG15th PPWGDokument29 SeitenASEAN BE Guidelines-Amended According To1st BABE TWG15th PPWGTarikNoch keine Bewertungen
- Bioavailability and BioequivalenceDokument81 SeitenBioavailability and BioequivalenceNiharika ModiNoch keine Bewertungen
- Bioavailability and BioequivalenceDokument39 SeitenBioavailability and Bioequivalenceنور الهدىNoch keine Bewertungen
- Comparing Generic and Innovator Drugs A Review ofDokument16 SeitenComparing Generic and Innovator Drugs A Review ofPrachi PandeyNoch keine Bewertungen
- Ad. BPPK 16Dokument119 SeitenAd. BPPK 16Sachin BagewadiNoch keine Bewertungen
- Bioavailability: by Abera JDokument59 SeitenBioavailability: by Abera JAbera JamboNoch keine Bewertungen
- Under Guidance of Mr. Mali K.K. (Assistant Professor) : 1 Yspm, YtcDokument41 SeitenUnder Guidance of Mr. Mali K.K. (Assistant Professor) : 1 Yspm, YtcSabiruddin Mirza DipuNoch keine Bewertungen
- BiopharmaceuticsDokument21 SeitenBiopharmaceuticsSilvy100% (1)
- The Orange BookDokument6 SeitenThe Orange Book50KMKDIVYA RAJPALNoch keine Bewertungen
- Bioavailability and Bieq Review ArticleDokument15 SeitenBioavailability and Bieq Review ArticlevinayNoch keine Bewertungen
- Pharmacy Technician Certification 8172019 ExamDokument115 SeitenPharmacy Technician Certification 8172019 Examfatdaddys100% (2)
- Guidance: in Vivo BioequivalenceDokument14 SeitenGuidance: in Vivo BioequivalenceAnonymous Se5IdneSpNoch keine Bewertungen
- Note For Guidance On The Investigation of Bioavailability & BioequivalenceDokument19 SeitenNote For Guidance On The Investigation of Bioavailability & BioequivalenceAhmed AliNoch keine Bewertungen
- THEORANGEBOOKDokument6 SeitenTHEORANGEBOOKMashiko DakhundaridzeNoch keine Bewertungen
- Bioequivalence StudiesDokument19 SeitenBioequivalence StudiesYaqeen Mutan100% (1)
- Bioavailability and BioequivalenceDokument6 SeitenBioavailability and BioequivalenceDharmesh PatelNoch keine Bewertungen
- Accelerated Stability Test of Ferrous Sulfate Tablets in WaterDokument27 SeitenAccelerated Stability Test of Ferrous Sulfate Tablets in WaterJeyma DacumosNoch keine Bewertungen
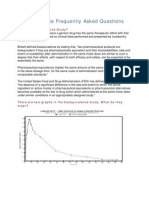
- Bioequivalence Frequently Asked Questions: What Is Bioequivalence Study?Dokument4 SeitenBioequivalence Frequently Asked Questions: What Is Bioequivalence Study?Ahmed HasanNoch keine Bewertungen
- Guideline For Bioequivalence Studies of Generic ProductsDokument23 SeitenGuideline For Bioequivalence Studies of Generic ProductschetanjmistryNoch keine Bewertungen
- Ceutics Study Guide QuizDokument6 SeitenCeutics Study Guide QuizJovanne D. BrownNoch keine Bewertungen
- Usp 1151Dokument10 SeitenUsp 1151Karnati PraveenaNoch keine Bewertungen
- Guidelines For Pharmaceutical Equivalence Requirements: Version 1.1Dokument9 SeitenGuidelines For Pharmaceutical Equivalence Requirements: Version 1.1rnd_holiNoch keine Bewertungen
- Purpose of BABE: Chu Yuan ShanDokument9 SeitenPurpose of BABE: Chu Yuan ShanJessie ChuNoch keine Bewertungen
- Requirement of Dissolution Test For f2 - Gastro Resistant TabletDokument17 SeitenRequirement of Dissolution Test For f2 - Gastro Resistant Tabletfad12345Noch keine Bewertungen
- Bioavailability and Bioequivalence StudiesDokument49 SeitenBioavailability and Bioequivalence StudiesvarishNoch keine Bewertungen
- Ayine Chap 4Dokument59 SeitenAyine Chap 4Abdi JifarNoch keine Bewertungen
- Therapeutic EquivalenceDokument2 SeitenTherapeutic EquivalenceAngel De la VictoriaNoch keine Bewertungen
- Bioavailability and BioequivalenceDokument23 SeitenBioavailability and BioequivalenceSaeed RashidNoch keine Bewertungen
- Therapeutic EquivalenceDokument2 SeitenTherapeutic EquivalenceyuppierajNoch keine Bewertungen
- BioequivalenceDokument30 SeitenBioequivalenceTejas PatelNoch keine Bewertungen
- AndaDokument26 SeitenAndaNagula NareshNoch keine Bewertungen
- Drug Stability for Pharmaceutical ScientistsVon EverandDrug Stability for Pharmaceutical ScientistsBewertung: 3.5 von 5 Sternen3.5/5 (3)
- Thesis About Mother Tongue Based EducationDokument8 SeitenThesis About Mother Tongue Based Educationkimstephenswashington100% (2)
- West Bengal DisaporaDokument301 SeitenWest Bengal Disaporaaman.4uNoch keine Bewertungen
- Encyclopedia of Clinical Pharmacy by Joseph T. DiPiroDokument958 SeitenEncyclopedia of Clinical Pharmacy by Joseph T. DiPiroAlex Pieces100% (1)
- Comparison of PubMed, Scopus, Web of Science, and Google Scholar - Strengths and WeaknessesDokument5 SeitenComparison of PubMed, Scopus, Web of Science, and Google Scholar - Strengths and WeaknessesMostafa AbdelrahmanNoch keine Bewertungen
- Nothemba Sitsheke, Acknowledge That Copying Someone Else's Assignment, or Part of It, IsDokument28 SeitenNothemba Sitsheke, Acknowledge That Copying Someone Else's Assignment, or Part of It, IsAshlyn LucasNoch keine Bewertungen
- 1 - Chapter 1 of Your Textbook Describes The Purposes of Nursing ResearDokument4 Seiten1 - Chapter 1 of Your Textbook Describes The Purposes of Nursing ResearHelping TutorsNoch keine Bewertungen
- Public Health Literature Review SampleDokument6 SeitenPublic Health Literature Review Sampleafmzywxfelvqoj100% (1)
- Research MethodolgyDokument4 SeitenResearch MethodolgyRizwan BalouchNoch keine Bewertungen
- High Quality Relationships Psychological Safety - Learning From Failures in Work OrganizationsDokument21 SeitenHigh Quality Relationships Psychological Safety - Learning From Failures in Work OrganizationsIacopo NicelliNoch keine Bewertungen
- Delta MethodDokument11 SeitenDelta MethodRaja Fawad ZafarNoch keine Bewertungen
- Fernando Da Vanci Cra Final DraftDokument6 SeitenFernando Da Vanci Cra Final Draftapi-462433002Noch keine Bewertungen
- For Sur SurDokument40 SeitenFor Sur SurJolie Mar ManceraNoch keine Bewertungen
- Excel For Statistical Data AnalysisDokument54 SeitenExcel For Statistical Data AnalysisLords PorseenaNoch keine Bewertungen
- Contribution of Tourism Industry in Indian Economy: An AnalysisDokument8 SeitenContribution of Tourism Industry in Indian Economy: An AnalysisHarsh GuptaNoch keine Bewertungen
- Anthropology Full All-ColourDokument28 SeitenAnthropology Full All-ColourLeinadNoch keine Bewertungen
- Chapter 4 000Dokument46 SeitenChapter 4 000Chandrahasa Reddy ThatimakulaNoch keine Bewertungen
- A Study On Training & Development in Kurnool: Yamaha Motors P.Chakrapani ReddyDokument7 SeitenA Study On Training & Development in Kurnool: Yamaha Motors P.Chakrapani ReddybagyaNoch keine Bewertungen
- Thesis RevDokument61 SeitenThesis RevJasond P. CapiliNoch keine Bewertungen
- SalDokument8 SeitenSalRobertoNoch keine Bewertungen
- Sample of Informed Consent FormDokument3 SeitenSample of Informed Consent FormHimekanoshikitaNoch keine Bewertungen
- Social Media Envy: How Experience Sharing On Social Networking Sites Drives Millennials' Aspirational Tourism ConsumptionDokument15 SeitenSocial Media Envy: How Experience Sharing On Social Networking Sites Drives Millennials' Aspirational Tourism ConsumptionMoch Aditya AkbarNoch keine Bewertungen
- Clinical Risk Management Systems 1Dokument11 SeitenClinical Risk Management Systems 1Dharmik ninama100% (4)
- Gnap 1995 Conducted A Survey On Teacher Workload Which Was Used in Exercise 16 of Chapter 5 A 3Dokument2 SeitenGnap 1995 Conducted A Survey On Teacher Workload Which Was Used in Exercise 16 of Chapter 5 A 3Charlotte0% (1)
- Benford's Law - WikipediaDokument7 SeitenBenford's Law - WikipediaBakari HamisiNoch keine Bewertungen
- Internal Audit Sampling by IIADokument11 SeitenInternal Audit Sampling by IIAAndi Tri Jati86% (7)
- AEC 12 - Q1 - 0503 - PS - Industry Analysis of Local BusinessesDokument45 SeitenAEC 12 - Q1 - 0503 - PS - Industry Analysis of Local BusinessesABM 11-10RAFER, FRANCHESCA KYLE S.Noch keine Bewertungen
- Interview Flashcards - QuizletDokument3 SeitenInterview Flashcards - QuizletKazuma SatouNoch keine Bewertungen
- Mapua Thesis Format 2020Dokument12 SeitenMapua Thesis Format 2020Synchelle CarmeloNoch keine Bewertungen
- Investigation of Pedestrian Safety A CasDokument11 SeitenInvestigation of Pedestrian Safety A CasElijah Jane RiveraNoch keine Bewertungen
- ENISA Cyber-Exercises Analysis Report-V1.0 PDFDokument32 SeitenENISA Cyber-Exercises Analysis Report-V1.0 PDFPaulo FelixNoch keine Bewertungen