Das könnte Ihnen auch gefallen
- Shoe Dog: A Memoir by the Creator of NikeVon EverandShoe Dog: A Memoir by the Creator of NikeBewertung: 4.5 von 5 Sternen4.5/5 (537)
- Exam III Study GuideDokument2 SeitenExam III Study GuideAnonymous rhUNqC1s0Noch keine Bewertungen
- The Yellow House: A Memoir (2019 National Book Award Winner)Von EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Bewertung: 4 von 5 Sternen4/5 (98)
- 1986 - CHA - HAL - Thermodyn Prop C1-C4 Ideal GasDokument68 Seiten1986 - CHA - HAL - Thermodyn Prop C1-C4 Ideal GasAnonymous rhUNqC1s0Noch keine Bewertungen
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeVon EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeBewertung: 4 von 5 Sternen4/5 (5794)
- Fukuyama Group - Group Meeting Problems 01/11/2013: T-BuliDokument4 SeitenFukuyama Group - Group Meeting Problems 01/11/2013: T-BuliAnonymous rhUNqC1s0Noch keine Bewertungen
- Fukuyama Group - Group Meeting Problems 07/17/2011: Low Yield Under Basic ConditionDokument3 SeitenFukuyama Group - Group Meeting Problems 07/17/2011: Low Yield Under Basic ConditionAnonymous rhUNqC1s0Noch keine Bewertungen
- The Little Book of Hygge: Danish Secrets to Happy LivingVon EverandThe Little Book of Hygge: Danish Secrets to Happy LivingBewertung: 3.5 von 5 Sternen3.5/5 (400)
- Fukuyama Group - Group Meeting Problems 09/07/2011: O N O Me MeDokument2 SeitenFukuyama Group - Group Meeting Problems 09/07/2011: O N O Me MeAnonymous rhUNqC1s0Noch keine Bewertungen
- Grit: The Power of Passion and PerseveranceVon EverandGrit: The Power of Passion and PerseveranceBewertung: 4 von 5 Sternen4/5 (588)
- Fukuyama Group - Group Meeting Problems 08/08/2011: T-Buok, N-Buli N-Buli (1.1 Eq)Dokument3 SeitenFukuyama Group - Group Meeting Problems 08/08/2011: T-Buok, N-Buli N-Buli (1.1 Eq)Anonymous rhUNqC1s0Noch keine Bewertungen
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureVon EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureBewertung: 4.5 von 5 Sternen4.5/5 (474)
- Fukuyama Group - Group Meeting Problems 09/05/2012: P-Tsoh (Cat.)Dokument2 SeitenFukuyama Group - Group Meeting Problems 09/05/2012: P-Tsoh (Cat.)Anonymous rhUNqC1s0Noch keine Bewertungen
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryVon EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryBewertung: 3.5 von 5 Sternen3.5/5 (231)
- Fukuyama Group - Group Meeting Problems 11/10/2012: PH BF KDokument3 SeitenFukuyama Group - Group Meeting Problems 11/10/2012: PH BF KAnonymous rhUNqC1s0Noch keine Bewertungen
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceVon EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceBewertung: 4 von 5 Sternen4/5 (895)
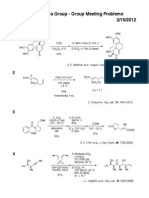
- Fukuyama Group - Group Meeting Problems 2/15/2012: Et Si Siet O D Thf-Hmpa - 78 °C 80% Et Si Siet Oh DDokument1 SeiteFukuyama Group - Group Meeting Problems 2/15/2012: Et Si Siet O D Thf-Hmpa - 78 °C 80% Et Si Siet Oh DAnonymous rhUNqC1s0Noch keine Bewertungen
- Team of Rivals: The Political Genius of Abraham LincolnVon EverandTeam of Rivals: The Political Genius of Abraham LincolnBewertung: 4.5 von 5 Sternen4.5/5 (234)
- Fukuyama Group - Group Meeting Problems 10/06/2012: J. Rodriguez Et Al., Org. Lett., 12, 4212 (2010)Dokument3 SeitenFukuyama Group - Group Meeting Problems 10/06/2012: J. Rodriguez Et Al., Org. Lett., 12, 4212 (2010)Anonymous rhUNqC1s0Noch keine Bewertungen
- Never Split the Difference: Negotiating As If Your Life Depended On ItVon EverandNever Split the Difference: Negotiating As If Your Life Depended On ItBewertung: 4.5 von 5 Sternen4.5/5 (838)
- Fukuyama Group - Group Meeting Problems 12/12/2012: CO Me NDokument2 SeitenFukuyama Group - Group Meeting Problems 12/12/2012: CO Me NAnonymous rhUNqC1s0Noch keine Bewertungen
- The Emperor of All Maladies: A Biography of CancerVon EverandThe Emperor of All Maladies: A Biography of CancerBewertung: 4.5 von 5 Sternen4.5/5 (271)
- Fukuyama Group - Group Meeting Problems 1/17/2012: Bicyclic CompoundDokument1 SeiteFukuyama Group - Group Meeting Problems 1/17/2012: Bicyclic CompoundAnonymous rhUNqC1s0Noch keine Bewertungen
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaVon EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaBewertung: 4.5 von 5 Sternen4.5/5 (266)
- Fukuyama Group - Group Meeting Problems 08/01/2012: Otes MeDokument3 SeitenFukuyama Group - Group Meeting Problems 08/01/2012: Otes MeAnonymous rhUNqC1s0Noch keine Bewertungen
- On Fire: The (Burning) Case for a Green New DealVon EverandOn Fire: The (Burning) Case for a Green New DealBewertung: 4 von 5 Sternen4/5 (74)
- Fukuyama Group - Group Meeting Problems 10/01/2011: N-Buli (2 Eq), - 20 °CDokument2 SeitenFukuyama Group - Group Meeting Problems 10/01/2011: N-Buli (2 Eq), - 20 °CAnonymous rhUNqC1s0Noch keine Bewertungen
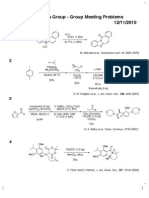
- Fukuyama Group - Group Meeting Problems 12/11/2010: T-BuomeDokument1 SeiteFukuyama Group - Group Meeting Problems 12/11/2010: T-BuomeAnonymous rhUNqC1s0Noch keine Bewertungen
- The Unwinding: An Inner History of the New AmericaVon EverandThe Unwinding: An Inner History of the New AmericaBewertung: 4 von 5 Sternen4/5 (45)
- Fukuyama Group - Group Meeting Problems 06/02/2010: O P N O Ome OmeDokument2 SeitenFukuyama Group - Group Meeting Problems 06/02/2010: O P N O Ome OmeAnonymous rhUNqC1s0Noch keine Bewertungen
- Astm E407Dokument21 SeitenAstm E407Chan UeiNianNoch keine Bewertungen
- Plant Design For Methanol Distillation Unit: February 2021Dokument51 SeitenPlant Design For Methanol Distillation Unit: February 2021maged1998100% (1)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersVon EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersBewertung: 4.5 von 5 Sternen4.5/5 (345)
- Exposure of Metals and Alloys by Alternate Immersion in Neutral 3.5 % Sodium Chloride SolutionDokument5 SeitenExposure of Metals and Alloys by Alternate Immersion in Neutral 3.5 % Sodium Chloride SolutionAliaNoch keine Bewertungen
- Metallography: Standard Terminology Relating ToDokument33 SeitenMetallography: Standard Terminology Relating To陳勉中Noch keine Bewertungen
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyVon EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyBewertung: 3.5 von 5 Sternen3.5/5 (2259)
- Laboratory Manual CHE102 Chemistry LabDokument44 SeitenLaboratory Manual CHE102 Chemistry Labnamita1276Noch keine Bewertungen
- Unit 4 Chemical Bonding & Molecular StructureDokument29 SeitenUnit 4 Chemical Bonding & Molecular StructureVighnesh0% (1)
- Schedule - 1 Classification of Industrial Units or Projects Based On Its Location and Impact On EnvironmentDokument8 SeitenSchedule - 1 Classification of Industrial Units or Projects Based On Its Location and Impact On EnvironmentFaruque UddinNoch keine Bewertungen
- Pharmaceutical Merits&Demerits of EmulsionDokument6 SeitenPharmaceutical Merits&Demerits of EmulsionShemaj GurchumaNoch keine Bewertungen
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreVon EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreBewertung: 4 von 5 Sternen4/5 (1090)
- 0041-En-2009-09 - Ventoplex Classifier Type CDokument4 Seiten0041-En-2009-09 - Ventoplex Classifier Type CJoão BaptistaNoch keine Bewertungen
- Dasteo N ButylDokument9 SeitenDasteo N ButylImtikhana KhofifahNoch keine Bewertungen
- M.Pharm I Semester: Gastroretentive Drug Delivery SystemsDokument58 SeitenM.Pharm I Semester: Gastroretentive Drug Delivery SystemsJAGADEESAN BALAJI100% (1)
- XI Ch1 Some Basic Concepts HssliveDokument2 SeitenXI Ch1 Some Basic Concepts HssliveJoshua Chacko Ponnachan100% (2)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Von EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Bewertung: 4.5 von 5 Sternen4.5/5 (121)
- 02 SpectroscopystratDokument3 Seiten02 SpectroscopystratDhiraj PatilNoch keine Bewertungen
- Showcase2015 CHE AlhajiriDokument1 SeiteShowcase2015 CHE AlhajiriAlex RichardNoch keine Bewertungen
- 634914904072057500Dokument35 Seiten634914904072057500Arjun HereNoch keine Bewertungen
- Redox Reactions Grade 11Dokument69 SeitenRedox Reactions Grade 11RahulNoch keine Bewertungen
- NMC Cathode Precursor Synthesis by Hydroxide Co-Precipitation Method: A Mini ReviewDokument8 SeitenNMC Cathode Precursor Synthesis by Hydroxide Co-Precipitation Method: A Mini Reviewwazzupal.yallNoch keine Bewertungen
- 045Dokument2 Seiten045Ghichajiwala_A_5413Noch keine Bewertungen
- Oxidation AllenDokument10 SeitenOxidation AllenAditi KharatNoch keine Bewertungen
- Succinic AcidDokument13 SeitenSuccinic AcidKaye Dimaano100% (1)
- To Quantitative ChemistryDokument37 SeitenTo Quantitative ChemistryVictor GuanNoch keine Bewertungen
- Lubri Oil Additives PDFDokument47 SeitenLubri Oil Additives PDFTejas PatelNoch keine Bewertungen
- Her Body and Other Parties: StoriesVon EverandHer Body and Other Parties: StoriesBewertung: 4 von 5 Sternen4/5 (821)
- PDEA Controlled Precursors and Essential ChemicalS ListDokument1 SeitePDEA Controlled Precursors and Essential ChemicalS ListJane PaladNoch keine Bewertungen
- Wet Storage Stain On Galvanized Steel PDFDokument8 SeitenWet Storage Stain On Galvanized Steel PDFmariustudoracheNoch keine Bewertungen
- Grade 10 Science (Chemistry Unit)Dokument12 SeitenGrade 10 Science (Chemistry Unit)Aastha SinghNoch keine Bewertungen
- Rita Maria Burguete Bacelar Marreiros FigueiraDokument203 SeitenRita Maria Burguete Bacelar Marreiros Figueiraenebravo12Noch keine Bewertungen
- C12SMR PhosphorylationDokument42 SeitenC12SMR PhosphorylationFelicia Ooi Ze EnNoch keine Bewertungen
- Activity 2-Solubility EquilibriumDokument5 SeitenActivity 2-Solubility Equilibriumalexandria heinleinNoch keine Bewertungen
- Sahan 2018 IOP Conf. Ser. - Mater. Sci. Eng. 345 012038 2Dokument8 SeitenSahan 2018 IOP Conf. Ser. - Mater. Sci. Eng. 345 012038 2pebrian sahputraNoch keine Bewertungen
- Bromoform Manufacturers, Phosphorus Tribromide, Diphenyl Acetonitrile.Dokument1 SeiteBromoform Manufacturers, Phosphorus Tribromide, Diphenyl Acetonitrile.Purecha GroupNoch keine Bewertungen
- It's Elemental: The Hidden Chemistry in EverythingVon EverandIt's Elemental: The Hidden Chemistry in EverythingBewertung: 4 von 5 Sternen4/5 (10)
- The Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactVon EverandThe Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactBewertung: 5 von 5 Sternen5/5 (5)