Das könnte Ihnen auch gefallen
- Shoe Dog: A Memoir by the Creator of NikeVon EverandShoe Dog: A Memoir by the Creator of NikeBewertung: 4.5 von 5 Sternen4.5/5 (537)
- Harvard System of ReferenceDokument7 SeitenHarvard System of Referencejaya1816Noch keine Bewertungen
- Grit: The Power of Passion and PerseveranceVon EverandGrit: The Power of Passion and PerseveranceBewertung: 4 von 5 Sternen4/5 (587)
- Mal LCDokument4 SeitenMal LCreborn007Noch keine Bewertungen
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceVon EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceBewertung: 4 von 5 Sternen4/5 (894)
- Evidence For DNA As The Hereditary MoleculeDokument2 SeitenEvidence For DNA As The Hereditary MoleculeShaezarah MohamudallyNoch keine Bewertungen
- The Yellow House: A Memoir (2019 National Book Award Winner)Von EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Bewertung: 4 von 5 Sternen4/5 (98)
- Ch10-1 Gen MaterialDokument36 SeitenCh10-1 Gen MaterialarcheologistsNoch keine Bewertungen
- The Little Book of Hygge: Danish Secrets to Happy LivingVon EverandThe Little Book of Hygge: Danish Secrets to Happy LivingBewertung: 3.5 von 5 Sternen3.5/5 (399)
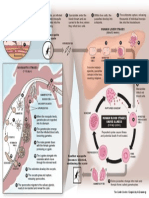
- Malaria Life Cycle ChartDokument1 SeiteMalaria Life Cycle ChartShaezarah MohamudallyNoch keine Bewertungen
- On Fire: The (Burning) Case for a Green New DealVon EverandOn Fire: The (Burning) Case for a Green New DealBewertung: 4 von 5 Sternen4/5 (73)
- Similarities Between Prokaryotic and Eukaryotic CellsDokument2 SeitenSimilarities Between Prokaryotic and Eukaryotic CellsShaezarah MohamudallyNoch keine Bewertungen
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeVon EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeBewertung: 4 von 5 Sternen4/5 (5794)
- Identifying Unknown Bacterium-GuideDokument13 SeitenIdentifying Unknown Bacterium-GuideShaezarah MohamudallyNoch keine Bewertungen
- Never Split the Difference: Negotiating As If Your Life Depended On ItVon EverandNever Split the Difference: Negotiating As If Your Life Depended On ItBewertung: 4.5 von 5 Sternen4.5/5 (838)
- Biochemical Composition of Fish - A PrimerDokument6 SeitenBiochemical Composition of Fish - A PrimerShaezarah MohamudallyNoch keine Bewertungen
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureVon EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureBewertung: 4.5 von 5 Sternen4.5/5 (474)
- Gram StainDokument1 SeiteGram StainShaezarah MohamudallyNoch keine Bewertungen
- Ascomycotina Class PPT 2014Dokument80 SeitenAscomycotina Class PPT 2014Shaezarah Mohamudally67% (3)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryVon EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryBewertung: 3.5 von 5 Sternen3.5/5 (231)
- MicropipetteDokument7 SeitenMicropipetteMike Serge Razafi0% (1)
- Characteristics of Fungi N ProtistDokument8 SeitenCharacteristics of Fungi N ProtistShaezarah MohamudallyNoch keine Bewertungen
- The Emperor of All Maladies: A Biography of CancerVon EverandThe Emperor of All Maladies: A Biography of CancerBewertung: 4.5 von 5 Sternen4.5/5 (271)
- Turbidity ScienceDokument26 SeitenTurbidity ScienceShaezarah MohamudallyNoch keine Bewertungen
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreVon EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreBewertung: 4 von 5 Sternen4/5 (1090)
- Fungus Lec NotesDokument8 SeitenFungus Lec NotesShaezarah MohamudallyNoch keine Bewertungen
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyVon EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyBewertung: 3.5 von 5 Sternen3.5/5 (2219)
- Malaria PDFDokument36 SeitenMalaria PDFYesi Novia AmbaraniNoch keine Bewertungen
- Team of Rivals: The Political Genius of Abraham LincolnVon EverandTeam of Rivals: The Political Genius of Abraham LincolnBewertung: 4.5 von 5 Sternen4.5/5 (234)
- Topic3 BIOL1030NRDokument11 SeitenTopic3 BIOL1030NRShaezarah MohamudallyNoch keine Bewertungen
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersVon EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersBewertung: 4.5 von 5 Sternen4.5/5 (344)
- Malaria Parasites and DiseaseDokument7 SeitenMalaria Parasites and DiseaseBalaji Rao NNoch keine Bewertungen
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaVon EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaBewertung: 4.5 von 5 Sternen4.5/5 (265)
- Diff Cent PDFDokument33 SeitenDiff Cent PDFShaezarah MohamudallyNoch keine Bewertungen
- Fungus Lec NotesDokument8 SeitenFungus Lec NotesShaezarah MohamudallyNoch keine Bewertungen
- Boltzmann DistributionDokument2 SeitenBoltzmann DistributionShaezarah MohamudallyNoch keine Bewertungen
- LieDokument1 SeiteLieShaezarah MohamudallyNoch keine Bewertungen
- Lecture of Organic ChemistryDokument13 SeitenLecture of Organic ChemistryNutDen R X NuttapongNoch keine Bewertungen
- Growth Curves of Micro-OrganismsDokument31 SeitenGrowth Curves of Micro-OrganismsShaezarah MohamudallyNoch keine Bewertungen
- The Unwinding: An Inner History of the New AmericaVon EverandThe Unwinding: An Inner History of the New AmericaBewertung: 4 von 5 Sternen4/5 (45)
- Cell Counting Using A HemacytometerDokument3 SeitenCell Counting Using A HemacytometerKris Cahyo Mulyatno100% (1)
- Microsoft PowerPoint - Bio1009ch13Dokument48 SeitenMicrosoft PowerPoint - Bio1009ch13Shaezarah MohamudallyNoch keine Bewertungen
- PolarityDokument1 SeitePolarityDeepakNoch keine Bewertungen
- ChemistryDokument9 SeitenChemistryShaezarah MohamudallyNoch keine Bewertungen
- RAlaminarflowcabinets PDFDokument4 SeitenRAlaminarflowcabinets PDFShaezarah MohamudallyNoch keine Bewertungen
- Lecture of Organic ChemistryDokument13 SeitenLecture of Organic ChemistryNutDen R X NuttapongNoch keine Bewertungen
- Audit Acq Pay Cycle & InventoryDokument39 SeitenAudit Acq Pay Cycle & InventoryVianney Claire RabeNoch keine Bewertungen
- Evolution of Bluetooth PDFDokument2 SeitenEvolution of Bluetooth PDFJuzerNoch keine Bewertungen
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Von EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Bewertung: 4.5 von 5 Sternen4.5/5 (119)
- Tigo Pesa Account StatementDokument7 SeitenTigo Pesa Account StatementPeter Ngicur Carthemi100% (1)
- ASMOPS 2016 - International Invitation PHILIPPINEDokument4 SeitenASMOPS 2016 - International Invitation PHILIPPINEMl Phil0% (3)
- SQL Guide AdvancedDokument26 SeitenSQL Guide AdvancedRustik2020Noch keine Bewertungen
- 4 Wheel ThunderDokument9 Seiten4 Wheel ThunderOlga Lucia Zapata SavaresseNoch keine Bewertungen
- Cold Rolled Steel Sections - Specification: Kenya StandardDokument21 SeitenCold Rolled Steel Sections - Specification: Kenya StandardPEng. Tech. Alvince KoreroNoch keine Bewertungen
- Skuld List of CorrespondentDokument351 SeitenSkuld List of CorrespondentKASHANNoch keine Bewertungen
- Maverick Brochure SMLDokument16 SeitenMaverick Brochure SMLmalaoui44Noch keine Bewertungen
- Non Circumvention Non Disclosure Agreement (TERENCE) SGDokument7 SeitenNon Circumvention Non Disclosure Agreement (TERENCE) SGLin ChrisNoch keine Bewertungen
- GATE ECE 2006 Actual PaperDokument33 SeitenGATE ECE 2006 Actual Paperkibrom atsbhaNoch keine Bewertungen
- Her Body and Other Parties: StoriesVon EverandHer Body and Other Parties: StoriesBewertung: 4 von 5 Sternen4/5 (821)
- CR Vs MarubeniDokument15 SeitenCR Vs MarubeniSudan TambiacNoch keine Bewertungen
- New Education Policy 2019Dokument55 SeitenNew Education Policy 2019Aakarshanam VenturesNoch keine Bewertungen
- DELcraFT Works CleanEra ProjectDokument31 SeitenDELcraFT Works CleanEra Projectenrico_britaiNoch keine Bewertungen
- Social Media Exposure and Its Perceived Impact On Students' Home-Based Tasks ProductivityDokument9 SeitenSocial Media Exposure and Its Perceived Impact On Students' Home-Based Tasks ProductivityJewel PascuaNoch keine Bewertungen
- Steam Turbine Theory and Practice by Kearton PDF 35Dokument4 SeitenSteam Turbine Theory and Practice by Kearton PDF 35KKDhNoch keine Bewertungen
- Estimation of Working CapitalDokument12 SeitenEstimation of Working CapitalsnehalgaikwadNoch keine Bewertungen
- To Introduce BgjgjgmyselfDokument2 SeitenTo Introduce Bgjgjgmyselflikith333Noch keine Bewertungen
- Casting Procedures and Defects GuideDokument91 SeitenCasting Procedures and Defects GuideJitender Reddy0% (1)
- Exp 8 - GPG - D12B - 74 PDFDokument4 SeitenExp 8 - GPG - D12B - 74 PDFPRATIKSHA WADIBHASMENoch keine Bewertungen
- Entity Level ControlsDokument45 SeitenEntity Level ControlsNiraj AlltimeNoch keine Bewertungen
- Axe Case Study - Call Me NowDokument6 SeitenAxe Case Study - Call Me NowvirgoashishNoch keine Bewertungen
- Acne Treatment Strategies and TherapiesDokument32 SeitenAcne Treatment Strategies and TherapiesdokterasadNoch keine Bewertungen
- Preventing and Mitigating COVID-19 at Work: Policy Brief 19 May 2021Dokument21 SeitenPreventing and Mitigating COVID-19 at Work: Policy Brief 19 May 2021Desy Fitriani SarahNoch keine Bewertungen
- ISO 9001:2015 Explained, Fourth Edition GuideDokument3 SeitenISO 9001:2015 Explained, Fourth Edition GuideiresendizNoch keine Bewertungen
- Rounded Scoodie Bobwilson123 PDFDokument3 SeitenRounded Scoodie Bobwilson123 PDFStefania MoldoveanuNoch keine Bewertungen
- PandPofCC (8th Edition)Dokument629 SeitenPandPofCC (8th Edition)Carlos Alberto CaicedoNoch keine Bewertungen
- Intro To Gas DynamicsDokument8 SeitenIntro To Gas DynamicsMSK65Noch keine Bewertungen
- Passenger E-Ticket: Booking DetailsDokument1 SeitePassenger E-Ticket: Booking Detailsvarun.agarwalNoch keine Bewertungen
- The European Journal of Applied Economics - Vol. 16 #2Dokument180 SeitenThe European Journal of Applied Economics - Vol. 16 #2Aleksandar MihajlovićNoch keine Bewertungen
- Coating and Drying Defects: Troubleshooting Operating ProblemsVon EverandCoating and Drying Defects: Troubleshooting Operating ProblemsBewertung: 5 von 5 Sternen5/5 (1)
- Is That a Fact?: Frauds, Quacks, and the Real Science of Everyday LifeVon EverandIs That a Fact?: Frauds, Quacks, and the Real Science of Everyday LifeBewertung: 4.5 von 5 Sternen4.5/5 (3)
- Science Goes Viral: Captivating Accounts of Science in Everyday LifeVon EverandScience Goes Viral: Captivating Accounts of Science in Everyday LifeBewertung: 5 von 5 Sternen5/5 (1)
- Chemistry for Breakfast: The Amazing Science of Everyday LifeVon EverandChemistry for Breakfast: The Amazing Science of Everyday LifeBewertung: 4.5 von 5 Sternen4.5/5 (14)
- The Periodic Table: A Very Short IntroductionVon EverandThe Periodic Table: A Very Short IntroductionBewertung: 4.5 von 5 Sternen4.5/5 (3)
- The Disappearing Spoon: And Other True Tales of Madness, Love, and the History of the World from the Periodic Table of the ElementsVon EverandThe Disappearing Spoon: And Other True Tales of Madness, Love, and the History of the World from the Periodic Table of the ElementsBewertung: 4 von 5 Sternen4/5 (146)
- The Elements We Live By: How Iron Helps Us Breathe, Potassium Lets Us See, and Other Surprising Superpowers of the Periodic TableVon EverandThe Elements We Live By: How Iron Helps Us Breathe, Potassium Lets Us See, and Other Surprising Superpowers of the Periodic TableBewertung: 3.5 von 5 Sternen3.5/5 (22)
- Essential Oil Chemistry Formulating Essential Oil Blends that Heal - Aldehyde - Ketone - Lactone: Healing with Essential OilVon EverandEssential Oil Chemistry Formulating Essential Oil Blends that Heal - Aldehyde - Ketone - Lactone: Healing with Essential OilBewertung: 5 von 5 Sternen5/5 (1)