Das könnte Ihnen auch gefallen
- Chromatography Theory, and Instrument CalibrationDokument26 SeitenChromatography Theory, and Instrument CalibrationSoledad ColmenarezNoch keine Bewertungen
- Chromatography PDFDokument24 SeitenChromatography PDFAleck HenryNoch keine Bewertungen
- Applications of Integration in Biomedical Science: by William T. Self UCF EXCEL Applications of CalculusDokument57 SeitenApplications of Integration in Biomedical Science: by William T. Self UCF EXCEL Applications of Calculusvijay2109Noch keine Bewertungen
- Chromatography 2Dokument70 SeitenChromatography 2NofrizalNoch keine Bewertungen
- Analytical Separation Science - 2015 - Samanidou - Basic LC Method Development and OptimizationDokument18 SeitenAnalytical Separation Science - 2015 - Samanidou - Basic LC Method Development and OptimizationShahinuzzamanAdaNoch keine Bewertungen
- 9.5.3 Chromatography: Biochemical Engineering: Principles and ConceptsDokument35 Seiten9.5.3 Chromatography: Biochemical Engineering: Principles and ConceptsKarthik MNoch keine Bewertungen
- Models - Echem.thin Layer ChronoamperometryDokument12 SeitenModels - Echem.thin Layer ChronoamperometryJaancaarloDiiazNoch keine Bewertungen
- Informe 1 AnalisisDokument23 SeitenInforme 1 AnalisisCarla ParraNoch keine Bewertungen
- Mangiapia 2016Dokument10 SeitenMangiapia 2016radouane chatitNoch keine Bewertungen
- 3-Final Exam (Feb.2009)Dokument23 Seiten3-Final Exam (Feb.2009)Manaal M. Ramadaan100% (1)
- Chromatography: California Association of Chemistry TeachersDokument5 SeitenChromatography: California Association of Chemistry TeachersLiyana HalimNoch keine Bewertungen
- Instrumental Analysis IDokument55 SeitenInstrumental Analysis IIbse ussoNoch keine Bewertungen
- HPLC 08Dokument13 SeitenHPLC 08Vikas SharmaNoch keine Bewertungen
- New Microsoft Office Word DocumentDokument28 SeitenNew Microsoft Office Word Documentbnar jNoch keine Bewertungen
- Principles of Gas Chromatography 2Dokument12 SeitenPrinciples of Gas Chromatography 2Enanahmed EnanNoch keine Bewertungen
- 26Dokument10 Seiten26Elsa VásquezNoch keine Bewertungen
- Gas-Liquid Chromatography PDFDokument24 SeitenGas-Liquid Chromatography PDFVyjayanthiNoch keine Bewertungen
- Chromatography: Theory & PracticeDokument29 SeitenChromatography: Theory & PracticeMaame Ama FrempongNoch keine Bewertungen
- Instrumental HPLCDokument7 SeitenInstrumental HPLCTok WanNoch keine Bewertungen
- Models - Chem.liquid Chromatography 1Dokument12 SeitenModels - Chem.liquid Chromatography 1Anoop Uchagawkar0% (1)
- Methods in Statistical KineticsDokument37 SeitenMethods in Statistical KineticsFreddy Rodrigo Navarro GajardoNoch keine Bewertungen
- 2007 Loukas BlendsDokument11 Seiten2007 Loukas BlendsJavier Ramos SpotifyNoch keine Bewertungen
- Chromatography Methods Part 1Dokument46 SeitenChromatography Methods Part 1hamidNoch keine Bewertungen
- Partition Chromatography PDFDokument11 SeitenPartition Chromatography PDFdania hjoujNoch keine Bewertungen
- Flow CytometryDokument7 SeitenFlow CytometryWNoch keine Bewertungen
- Molecules 25 06020 v3Dokument25 SeitenMolecules 25 06020 v3Dinda Dwi SeptianiNoch keine Bewertungen
- Principles of Gas ChromatographyDokument17 SeitenPrinciples of Gas ChromatographyAtef LasheenNoch keine Bewertungen
- Biomedical Engineering Challenges: A Chemical Engineering InsightVon EverandBiomedical Engineering Challenges: A Chemical Engineering InsightNoch keine Bewertungen
- $ State and Parameter Estimation in Chemical and Biochemical Processes - A TutorialDokument18 Seiten$ State and Parameter Estimation in Chemical and Biochemical Processes - A TutorialsamandondonNoch keine Bewertungen
- Analytical Characterization of BiotherapeuticsVon EverandAnalytical Characterization of BiotherapeuticsJennie R. LillNoch keine Bewertungen
- Chapter 28 Liquid ChromatographyDokument32 SeitenChapter 28 Liquid ChromatographyHenrique CostaNoch keine Bewertungen
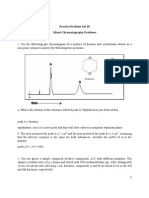
- Practice Problem Set Mixed Chromatography QuestionsDokument14 SeitenPractice Problem Set Mixed Chromatography QuestionsMelinda AndersonNoch keine Bewertungen
- Immunologhy Lab Report 3 - MURAT GENÇ-2021652244Dokument10 SeitenImmunologhy Lab Report 3 - MURAT GENÇ-2021652244Murat GençNoch keine Bewertungen
- Kromatografi KolomDokument40 SeitenKromatografi KolomghinaNoch keine Bewertungen
- Neighbour Lists in Smoothed Particle HydrodynamicsDokument17 SeitenNeighbour Lists in Smoothed Particle HydrodynamicsAditya BhosaleNoch keine Bewertungen
- An Overview of Recent Advances in HPLC InstrumentationDokument16 SeitenAn Overview of Recent Advances in HPLC Instrumentationlovina candra kiranaNoch keine Bewertungen
- Calculation of Membrane-Water Partition CoefficientDokument10 SeitenCalculation of Membrane-Water Partition CoefficientNickolay Melik-NubarovNoch keine Bewertungen
- HPLC Lab ManualDokument7 SeitenHPLC Lab ManualRakesh Kumar ChaudharyNoch keine Bewertungen
- ChromatographyDokument19 SeitenChromatographyppj25945Noch keine Bewertungen
- Reactive Oxygen Species: Signaling Between Hierarchical Levels in PlantsVon EverandReactive Oxygen Species: Signaling Between Hierarchical Levels in PlantsFranz-Josef SchmittNoch keine Bewertungen
- Application of MachineDokument21 SeitenApplication of MachineSengatan LebahNoch keine Bewertungen
- 03MathematicalModellingOfChemicalProcesses - Doc 0Dokument12 Seiten03MathematicalModellingOfChemicalProcesses - Doc 0Anonymous P1iMibNoch keine Bewertungen
- High Performance Liquid Chromatography (HPLC)Dokument9 SeitenHigh Performance Liquid Chromatography (HPLC)Secret ManNoch keine Bewertungen
- Flow in 3D Packed Bed Reactors COMSOLDokument10 SeitenFlow in 3D Packed Bed Reactors COMSOLBenedictEkowPrahNoch keine Bewertungen
- Practical Flow Cytometry - M.Sc. 2nd Sem Immuno - FinalDokument5 SeitenPractical Flow Cytometry - M.Sc. 2nd Sem Immuno - FinalAnusua RoyNoch keine Bewertungen
- Information Given in These Slides Are, Either in Part or All, Recollection From The FollowingsDokument52 SeitenInformation Given in These Slides Are, Either in Part or All, Recollection From The FollowingsHaile KassaNoch keine Bewertungen
- Chromatography PrintDokument30 SeitenChromatography PrintAmmad FazilNoch keine Bewertungen
- Lle Design PaperDokument14 SeitenLle Design PaperRamesh ReddyNoch keine Bewertungen
- An Introduction To Analytical Method Development For Pharmaceutical FormulationsDokument45 SeitenAn Introduction To Analytical Method Development For Pharmaceutical Formulationsapi-19786321100% (2)
- Experimental and Theoretical Optical properties of β-carotene in oleic acid solutionDokument14 SeitenExperimental and Theoretical Optical properties of β-carotene in oleic acid solutionrubensufpaNoch keine Bewertungen
- Biological Networks in Metabolic P Systems: Vincenzo Manca, Luca BiancoDokument10 SeitenBiological Networks in Metabolic P Systems: Vincenzo Manca, Luca Biancoapi-253266324Noch keine Bewertungen
- Jeremiah John Samontina - Activity 1Dokument3 SeitenJeremiah John Samontina - Activity 1jeremiah john samontinaNoch keine Bewertungen
- UNIT: SpectrophotometryDokument15 SeitenUNIT: SpectrophotometrybiddyusmcNoch keine Bewertungen
- Ultra Performance Liquid ChromatographyDokument8 SeitenUltra Performance Liquid ChromatographyQudsia RehmanNoch keine Bewertungen
- Shiv Aram An PaperDokument6 SeitenShiv Aram An Paperblabla9292Noch keine Bewertungen
- CHEM4330L6Dokument33 SeitenCHEM4330L6justinhadinata283Noch keine Bewertungen
- Laminar Flow and Diffusion in A MicrochannelDokument5 SeitenLaminar Flow and Diffusion in A MicrochannelBryan FonsecaNoch keine Bewertungen
- CHM510-e - LaboratoryManual Sem October 2020-Februari2021Dokument10 SeitenCHM510-e - LaboratoryManual Sem October 2020-Februari2021Amirah AzlanNoch keine Bewertungen
- Mass EnergyDokument64 SeitenMass EnergyhlvijaykumarNoch keine Bewertungen
- Selected Constants: Oxidation–Reduction Potentials of Inorganic Substances in Aqueous SolutionVon EverandSelected Constants: Oxidation–Reduction Potentials of Inorganic Substances in Aqueous SolutionNoch keine Bewertungen
- Bio in Stu MentationDokument68 SeitenBio in Stu Mentationprathaps1987Noch keine Bewertungen
- 08 Chapter3 PDFDokument34 Seiten08 Chapter3 PDFprathaps1987Noch keine Bewertungen
- Chemical Reactor Selection and Design: Rajesh Kumar Bhagat B.E. (CHE) 4 YearDokument6 SeitenChemical Reactor Selection and Design: Rajesh Kumar Bhagat B.E. (CHE) 4 Yearprathaps1987Noch keine Bewertungen
- Chap8 BioreactorDokument16 SeitenChap8 Bioreactorprathaps1987Noch keine Bewertungen
- Experiment 6 Thin Layer Chromatographic Separation and Identification of Amino AcidsDokument4 SeitenExperiment 6 Thin Layer Chromatographic Separation and Identification of Amino Acidsprathaps1987Noch keine Bewertungen
- Mendelian GeneticsDokument22 SeitenMendelian Geneticsprathaps1987Noch keine Bewertungen
- Experiment 6 Thin Layer Chromatographic Separation and Identification of Amino AcidsDokument4 SeitenExperiment 6 Thin Layer Chromatographic Separation and Identification of Amino Acidsprathaps1987Noch keine Bewertungen
- For IntroductionDokument10 SeitenFor Introductionprathaps1987Noch keine Bewertungen
- A Review of Rapid Pro To Typing Techniques For Tissue EngineeringDokument30 SeitenA Review of Rapid Pro To Typing Techniques For Tissue EngineeringEduardo SandovalNoch keine Bewertungen
- SD ArticleDokument2 SeitenSD Articleprathaps1987Noch keine Bewertungen
- 4 Kinetis ModelDokument12 Seiten4 Kinetis Modelprathaps1987Noch keine Bewertungen
- Drug Kinetics Correct One PDFDokument10 SeitenDrug Kinetics Correct One PDFprathaps1987Noch keine Bewertungen
- Full Idea PDFDokument8 SeitenFull Idea PDFprathaps1987Noch keine Bewertungen
- Full Idea PDFDokument8 SeitenFull Idea PDFprathaps1987Noch keine Bewertungen
- Tissue Engg Rapid PrototypingDokument11 SeitenTissue Engg Rapid Prototypingprathaps1987Noch keine Bewertungen
- 3D PrintingDokument8 Seiten3D Printingprathaps1987Noch keine Bewertungen
- Tissue Engg Rapid PrototypingDokument11 SeitenTissue Engg Rapid Prototypingprathaps1987Noch keine Bewertungen
- 1103 6177Dokument26 Seiten1103 6177prathaps1987Noch keine Bewertungen
- Covalent and Ionic BondingDokument22 SeitenCovalent and Ionic BondingBishan MeghaniNoch keine Bewertungen
- Pharmacuetics For The Students of Pharmacy Technicians (Category-B)Dokument45 SeitenPharmacuetics For The Students of Pharmacy Technicians (Category-B)Naina's kitchencookingvediosNoch keine Bewertungen
- EE145 HMWK 1 SolDokument11 SeitenEE145 HMWK 1 SolNuwan SameeraNoch keine Bewertungen
- SL MC Test s2 Models of Bonding - Structure (Second Test)Dokument7 SeitenSL MC Test s2 Models of Bonding - Structure (Second Test)Amira Selpa KhairunnisaNoch keine Bewertungen
- Mechanical Behaviour and Testing of MaterialsDokument811 SeitenMechanical Behaviour and Testing of MaterialsMohammed AbdulkedirNoch keine Bewertungen
- F 4 ScienceyearlyplanDokument25 SeitenF 4 ScienceyearlyplanFaridah EsaNoch keine Bewertungen
- Laynes - Chapter 10 GasesDokument142 SeitenLaynes - Chapter 10 GasescharleneNoch keine Bewertungen
- Pharmaceutical SciencesDokument9 SeitenPharmaceutical SciencesJames HornerNoch keine Bewertungen
- Ionic EquilibriumDokument24 SeitenIonic EquilibriumRaju SinghNoch keine Bewertungen
- 3 - Determining Activity SeriesDokument5 Seiten3 - Determining Activity Seriescarter0% (2)
- Elements in Period 3Dokument13 SeitenElements in Period 3FAthiyah Abdul RahimNoch keine Bewertungen
- 1 s2.0 S1383586623007190 MainDokument24 Seiten1 s2.0 S1383586623007190 MainDaniel MontalvoNoch keine Bewertungen
- Chemistry Paper 1 Revision Mat - Atomic StructureDokument2 SeitenChemistry Paper 1 Revision Mat - Atomic StructurekashificetNoch keine Bewertungen
- Design and Synthesis 2D Coordination Networks - Crcl2 (Pyz) 2 and CR (Oso2Ch3) 2 (Pyz) 2 (Pyz Pyrazine) Framework MagnetsDokument11 SeitenDesign and Synthesis 2D Coordination Networks - Crcl2 (Pyz) 2 and CR (Oso2Ch3) 2 (Pyz) 2 (Pyz Pyrazine) Framework MagnetsMercyJatindroNoch keine Bewertungen
- Lesson Plan Chemical BondingDokument11 SeitenLesson Plan Chemical BondingNelda100% (2)
- Unit 9 - Metals and Their Compounds Teacher VersionDokument29 SeitenUnit 9 - Metals and Their Compounds Teacher VersionAmadu sallieuNoch keine Bewertungen
- UntitledDokument422 SeitenUntitledAbanti MitraNoch keine Bewertungen
- Fundamentals of Polymer ChemistryDokument48 SeitenFundamentals of Polymer ChemistryRUSTSHIELD Indonesia100% (1)
- Monday 20 May 2019 - Morning: AS Level Chemistry ADokument12 SeitenMonday 20 May 2019 - Morning: AS Level Chemistry Asemirah anthonyNoch keine Bewertungen
- Masterton Odd No - AnswersDokument257 SeitenMasterton Odd No - Answers박시후Noch keine Bewertungen
- Applied Electronics I. HandoutDokument18 SeitenApplied Electronics I. HandoutzemichaelNoch keine Bewertungen
- Gromacs Tutorial 2 (Protein) PDFDokument19 SeitenGromacs Tutorial 2 (Protein) PDFEliasSMonteiroFilhoNoch keine Bewertungen
- What Are Negative Ions?Dokument4 SeitenWhat Are Negative Ions?Kedar KelkarNoch keine Bewertungen
- Madaraka F3 Discussion QuestionsDokument10 SeitenMadaraka F3 Discussion Questionsshad kojwangNoch keine Bewertungen
- Physical Science - CH 11Dokument5 SeitenPhysical Science - CH 11suhughes100% (5)
- Better Amine System Operations Through Chemical Analysis - LRGCC 2013 Finalrev2 PDFDokument32 SeitenBetter Amine System Operations Through Chemical Analysis - LRGCC 2013 Finalrev2 PDFShashank N Shah100% (2)
- DFGHJKLDokument102 SeitenDFGHJKLLucas MarchiniNoch keine Bewertungen
- Kimia JWP Bab 3Dokument23 SeitenKimia JWP Bab 3CHA ZI YU MoeNoch keine Bewertungen
- 84 - Werner's Theory of Coordination CompoundsDokument5 Seiten84 - Werner's Theory of Coordination CompoundsSyed HusamNoch keine Bewertungen
- MUCLecture 2022 51954485Dokument21 SeitenMUCLecture 2022 51954485Mr AliNoch keine Bewertungen