Das könnte Ihnen auch gefallen
- Grit: The Power of Passion and PerseveranceVon EverandGrit: The Power of Passion and PerseveranceBewertung: 4 von 5 Sternen4/5 (588)
- Abstracts For The 27th Annual Scientific Meeting of The Society For Immunotherapy of Cancer (SITC) PDFDokument71 SeitenAbstracts For The 27th Annual Scientific Meeting of The Society For Immunotherapy of Cancer (SITC) PDFhigginscribdNoch keine Bewertungen
- The Yellow House: A Memoir (2019 National Book Award Winner)Von EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Bewertung: 4 von 5 Sternen4/5 (98)
- ELP Language Biography Checklists For Young Learners enDokument8 SeitenELP Language Biography Checklists For Young Learners enhigginscribdNoch keine Bewertungen
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeVon EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeBewertung: 4 von 5 Sternen4/5 (5795)
- ALTE CEFR Speaking Grid Tests2014Dokument11 SeitenALTE CEFR Speaking Grid Tests2014higginscribdNoch keine Bewertungen
- Never Split the Difference: Negotiating As If Your Life Depended On ItVon EverandNever Split the Difference: Negotiating As If Your Life Depended On ItBewertung: 4.5 von 5 Sternen4.5/5 (838)
- ELP Language Biography Plurilingual Profile enDokument5 SeitenELP Language Biography Plurilingual Profile enhigginscribdNoch keine Bewertungen
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceVon EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceBewertung: 4 von 5 Sternen4/5 (895)
- ELP What Is A Portfolio ENDokument1 SeiteELP What Is A Portfolio ENhigginscribdNoch keine Bewertungen
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersVon EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersBewertung: 4.5 von 5 Sternen4.5/5 (345)
- Praepositiones LatinaeDokument1 SeitePraepositiones LatinaeMagister Optimus MaximusNoch keine Bewertungen
- Shoe Dog: A Memoir by the Creator of NikeVon EverandShoe Dog: A Memoir by the Creator of NikeBewertung: 4.5 von 5 Sternen4.5/5 (537)
- Catalog Sep 2014Dokument14 SeitenCatalog Sep 2014higginscribdNoch keine Bewertungen
- ELP Language Biography Generic Checklists enDokument18 SeitenELP Language Biography Generic Checklists enhigginscribdNoch keine Bewertungen
- The Little Book of Hygge: Danish Secrets to Happy LivingVon EverandThe Little Book of Hygge: Danish Secrets to Happy LivingBewertung: 3.5 von 5 Sternen3.5/5 (400)
- Luraghi Narrog 4 Luraghi-LibreDokument76 SeitenLuraghi Narrog 4 Luraghi-LibrehigginscribdNoch keine Bewertungen
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureVon EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureBewertung: 4.5 von 5 Sternen4.5/5 (474)
- 02.04.quiz 1 SolutionsDokument1 Seite02.04.quiz 1 SolutionshigginscribdNoch keine Bewertungen
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryVon EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryBewertung: 3.5 von 5 Sternen3.5/5 (231)
- What Are Complex Systems?Dokument18 SeitenWhat Are Complex Systems?higginscribdNoch keine Bewertungen
- On Fire: The (Burning) Case for a Green New DealVon EverandOn Fire: The (Burning) Case for a Green New DealBewertung: 4 von 5 Sternen4/5 (74)
- Differential Equations: Some Notes On Terminology: DX DT F (X)Dokument4 SeitenDifferential Equations: Some Notes On Terminology: DX DT F (X)higginscribdNoch keine Bewertungen
- The Emperor of All Maladies: A Biography of CancerVon EverandThe Emperor of All Maladies: A Biography of CancerBewertung: 4.5 von 5 Sternen4.5/5 (271)
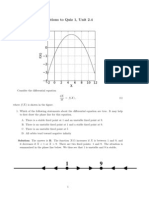
- Solutions To Quiz 2, Unit 2.4Dokument1 SeiteSolutions To Quiz 2, Unit 2.4higginscribdNoch keine Bewertungen
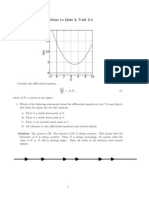
- Solutions To Quiz 1, Unit 2.2Dokument1 SeiteSolutions To Quiz 1, Unit 2.2higginscribdNoch keine Bewertungen
- 02.05AlgorithmicSolutions Summary 2Dokument3 Seiten02.05AlgorithmicSolutions Summary 2higginscribdNoch keine Bewertungen
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaVon EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaBewertung: 4.5 von 5 Sternen4.5/5 (266)
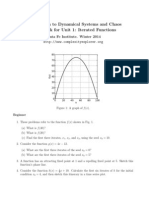
- Introduction To Dynamical Systems and Chaos Homework For Unit 1: Iterated FunctionsDokument2 SeitenIntroduction To Dynamical Systems and Chaos Homework For Unit 1: Iterated FunctionshigginscribdNoch keine Bewertungen
- The Unwinding: An Inner History of the New AmericaVon EverandThe Unwinding: An Inner History of the New AmericaBewertung: 4 von 5 Sternen4/5 (45)
- Self-Organization and Cooperation in Social SystemsDokument29 SeitenSelf-Organization and Cooperation in Social SystemshigginscribdNoch keine Bewertungen
- Team of Rivals: The Political Genius of Abraham LincolnVon EverandTeam of Rivals: The Political Genius of Abraham LincolnBewertung: 4.5 von 5 Sternen4.5/5 (234)
- Cellular Automata Are Idealized Models of Complex SystemsDokument58 SeitenCellular Automata Are Idealized Models of Complex SystemshigginscribdNoch keine Bewertungen
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyVon EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyBewertung: 3.5 von 5 Sternen3.5/5 (2259)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreVon EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreBewertung: 4 von 5 Sternen4/5 (1091)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Von EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Bewertung: 4.5 von 5 Sternen4.5/5 (121)
- Her Body and Other Parties: StoriesVon EverandHer Body and Other Parties: StoriesBewertung: 4 von 5 Sternen4/5 (821)