Das könnte Ihnen auch gefallen
- Syncope (fainting) causes, symptoms and treatmentDokument7 SeitenSyncope (fainting) causes, symptoms and treatmentWira SentanuNoch keine Bewertungen
- HomeopathyDokument46 SeitenHomeopathyWira SentanuNoch keine Bewertungen
- Nepal: Federal Democratic Republic of NepalDokument52 SeitenNepal: Federal Democratic Republic of NepalWira SentanuNoch keine Bewertungen
- History: For Other Uses of "Gill", SeeDokument6 SeitenHistory: For Other Uses of "Gill", SeeWira SentanuNoch keine Bewertungen
- Aquatic Ecosystem GuideDokument7 SeitenAquatic Ecosystem GuideWira SentanuNoch keine Bewertungen
- SanskritDokument25 SeitenSanskritWira SentanuNoch keine Bewertungen
- Biology: For Other Uses, SeeDokument12 SeitenBiology: For Other Uses, SeeWira SentanuNoch keine Bewertungen
- ReptileDokument31 SeitenReptileWira SentanuNoch keine Bewertungen
- Xan ThomaDokument4 SeitenXan ThomaWira SentanuNoch keine Bewertungen
- Discover the secrets of whiskersDokument12 SeitenDiscover the secrets of whiskersWira SentanuNoch keine Bewertungen
- Fish Evolution and AnatomyDokument24 SeitenFish Evolution and AnatomyWira SentanuNoch keine Bewertungen
- Koi Fish GuideDokument10 SeitenKoi Fish GuideWira SentanuNoch keine Bewertungen
- CatfishDokument16 SeitenCatfishWira SentanuNoch keine Bewertungen
- MammalDokument48 SeitenMammalWira SentanuNoch keine Bewertungen
- Tokugawa YashinobuDokument9 SeitenTokugawa YashinobuWira SentanuNoch keine Bewertungen
- HELLP SyndromeDokument6 SeitenHELLP SyndromeWira SentanuNoch keine Bewertungen
- ShogunDokument7 SeitenShogunWira SentanuNoch keine Bewertungen
- MicroorganismDokument23 SeitenMicroorganismWira SentanuNoch keine Bewertungen
- Conti GuityDokument5 SeitenConti GuityWira SentanuNoch keine Bewertungen
- CoughDokument8 SeitenCoughWira SentanuNoch keine Bewertungen
- Sarcoptes ScabieiDokument4 SeitenSarcoptes ScabieiWira SentanuNoch keine Bewertungen
- TropismDokument3 SeitenTropismWira SentanuNoch keine Bewertungen
- BloodDokument28 SeitenBloodWira SentanuNoch keine Bewertungen
- ScabiesDokument15 SeitenScabiesWira SentanuNoch keine Bewertungen
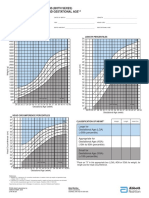
- Gestational AgeDokument9 SeitenGestational AgeWira SentanuNoch keine Bewertungen
- Intrauterine Growth RestrictionDokument9 SeitenIntrauterine Growth RestrictionWira SentanuNoch keine Bewertungen
- AristotleDokument56 SeitenAristotleWira SentanuNoch keine Bewertungen
- HeliotropismDokument3 SeitenHeliotropismWira SentanuNoch keine Bewertungen
- OrganismDokument17 SeitenOrganismWira SentanuNoch keine Bewertungen
- PhotosensitivityDokument3 SeitenPhotosensitivityWira SentanuNoch keine Bewertungen
- Shoe Dog: A Memoir by the Creator of NikeVon EverandShoe Dog: A Memoir by the Creator of NikeBewertung: 4.5 von 5 Sternen4.5/5 (537)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeVon EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeBewertung: 4 von 5 Sternen4/5 (5794)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceVon EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceBewertung: 4 von 5 Sternen4/5 (890)
- The Yellow House: A Memoir (2019 National Book Award Winner)Von EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Bewertung: 4 von 5 Sternen4/5 (98)
- The Little Book of Hygge: Danish Secrets to Happy LivingVon EverandThe Little Book of Hygge: Danish Secrets to Happy LivingBewertung: 3.5 von 5 Sternen3.5/5 (399)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryVon EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryBewertung: 3.5 von 5 Sternen3.5/5 (231)
- Never Split the Difference: Negotiating As If Your Life Depended On ItVon EverandNever Split the Difference: Negotiating As If Your Life Depended On ItBewertung: 4.5 von 5 Sternen4.5/5 (838)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureVon EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureBewertung: 4.5 von 5 Sternen4.5/5 (474)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersVon EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersBewertung: 4.5 von 5 Sternen4.5/5 (344)
- Grit: The Power of Passion and PerseveranceVon EverandGrit: The Power of Passion and PerseveranceBewertung: 4 von 5 Sternen4/5 (587)
- On Fire: The (Burning) Case for a Green New DealVon EverandOn Fire: The (Burning) Case for a Green New DealBewertung: 4 von 5 Sternen4/5 (73)
- The Emperor of All Maladies: A Biography of CancerVon EverandThe Emperor of All Maladies: A Biography of CancerBewertung: 4.5 von 5 Sternen4.5/5 (271)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaVon EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaBewertung: 4.5 von 5 Sternen4.5/5 (265)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreVon EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreBewertung: 4 von 5 Sternen4/5 (1090)
- Team of Rivals: The Political Genius of Abraham LincolnVon EverandTeam of Rivals: The Political Genius of Abraham LincolnBewertung: 4.5 von 5 Sternen4.5/5 (234)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyVon EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyBewertung: 3.5 von 5 Sternen3.5/5 (2219)
- The Unwinding: An Inner History of the New AmericaVon EverandThe Unwinding: An Inner History of the New AmericaBewertung: 4 von 5 Sternen4/5 (45)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Von EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Bewertung: 4.5 von 5 Sternen4.5/5 (119)
- Her Body and Other Parties: StoriesVon EverandHer Body and Other Parties: StoriesBewertung: 4 von 5 Sternen4/5 (821)
- Recall MRCP 1 Jan 2014Dokument20 SeitenRecall MRCP 1 Jan 2014Mohamed Amr SalamaNoch keine Bewertungen
- Carlos Vascular PharmacologyDokument8 SeitenCarlos Vascular PharmacologyAfif AuliyaNoch keine Bewertungen
- Postnatal CareDokument19 SeitenPostnatal Carejomarie gamiao0% (1)
- Chapter 15Dokument27 SeitenChapter 15Christian JovelNoch keine Bewertungen
- Instructions Cyborg Beast Updated 6-26-14Dokument18 SeitenInstructions Cyborg Beast Updated 6-26-14fredyugaNoch keine Bewertungen
- Patient Evaluation and Wound Assessment: Key Practice PointsDokument6 SeitenPatient Evaluation and Wound Assessment: Key Practice PointsFofiuNoch keine Bewertungen
- An Efficacy and Safety of Nanofractional Radiofrequency For The Treatment of Striae AlbaDokument7 SeitenAn Efficacy and Safety of Nanofractional Radiofrequency For The Treatment of Striae AlbaDomenico NarcisoNoch keine Bewertungen
- Cutaneous Lupus Erythematosus Diagnosis and Treatment1Dokument14 SeitenCutaneous Lupus Erythematosus Diagnosis and Treatment1Crystine SinatraNoch keine Bewertungen
- MAPO Index For Risk Assessment of Patient Manual Handling in Hospital Wards: A Validation StudyDokument17 SeitenMAPO Index For Risk Assessment of Patient Manual Handling in Hospital Wards: A Validation StudySebastian LoperaNoch keine Bewertungen
- Lubchenco Curve PDFDokument1 SeiteLubchenco Curve PDFWarren Lie25% (4)
- Camp Rap-A-Hope - 2013 Annual ReportDokument20 SeitenCamp Rap-A-Hope - 2013 Annual ReportWest SandersNoch keine Bewertungen
- Clinical Biomechanics in Implant DentistryDokument36 SeitenClinical Biomechanics in Implant DentistryMahadevan Ravichandran100% (4)
- Hemostasis and Blood Coagulation PDFDokument23 SeitenHemostasis and Blood Coagulation PDFLaura Chandra100% (3)
- Abc Abuso 1 PDFDokument6 SeitenAbc Abuso 1 PDFMDMNoch keine Bewertungen
- Dubuque Management of Clinical Alarms SystemsDokument6 SeitenDubuque Management of Clinical Alarms SystemsAhmed El KadyNoch keine Bewertungen
- Dorothy Johnson (Report)Dokument24 SeitenDorothy Johnson (Report)mlbonthelineNoch keine Bewertungen
- Cleft Lip and Palate CareDokument16 SeitenCleft Lip and Palate CareIsmail LubisNoch keine Bewertungen
- Child PoliciesDokument4 SeitenChild PoliciesMon DeNoch keine Bewertungen
- Slide Tentang Bite Plane PDFDokument3 SeitenSlide Tentang Bite Plane PDFRahmat HidayatNoch keine Bewertungen
- Assignment 1 Introducing HealthDokument13 SeitenAssignment 1 Introducing Health177 Gede Agus Purna YogaNoch keine Bewertungen
- Presented by Dr. K. M. Rukhshad Asif Zaman Khan Department of Orthodontics and Dentofacial OrthopaedicsDokument21 SeitenPresented by Dr. K. M. Rukhshad Asif Zaman Khan Department of Orthodontics and Dentofacial OrthopaedicsRukshad Asif Jaman KhanNoch keine Bewertungen
- Newborn Resuscitation StepsDokument34 SeitenNewborn Resuscitation StepsNishaThakuri100% (2)
- Dental Caries CaseDokument5 SeitenDental Caries CaseEerrmmaa Ssaarrell Rroommaann PpaarraaNoch keine Bewertungen
- Integrative Literature ReviewDokument17 SeitenIntegrative Literature Reviewapi-426049026Noch keine Bewertungen
- Jewish Standard Readers' Choice 2016Dokument80 SeitenJewish Standard Readers' Choice 2016New Jersey Jewish StandardNoch keine Bewertungen
- Chenhua Yan, MD: ChinaDokument34 SeitenChenhua Yan, MD: ChinaaymenNoch keine Bewertungen
- NIMS Electronic Telephone Exchange Provisional NumbersDokument13 SeitenNIMS Electronic Telephone Exchange Provisional NumbersSumant BhardvajNoch keine Bewertungen
- Skull Fracture vs. Accessory Sutures: How to Tell the DifferenceDokument6 SeitenSkull Fracture vs. Accessory Sutures: How to Tell the Differencemohit372Noch keine Bewertungen
- F. Daftar Pustaka (KIAN Lukman N 3)Dokument7 SeitenF. Daftar Pustaka (KIAN Lukman N 3)Lukman SulistiyadiNoch keine Bewertungen
- Managing Sickle Cell Anemia: A Nursing GuideDokument48 SeitenManaging Sickle Cell Anemia: A Nursing Guidejomcy0% (2)