Das könnte Ihnen auch gefallen
- Production FormalinDokument5 SeitenProduction FormalinVirginia Rosales Olmos0% (1)
- Modeling of Industrial Formaldehyde AbsorbersDokument19 SeitenModeling of Industrial Formaldehyde AbsorbersNguyen Thanh SangNoch keine Bewertungen
- Rate-Based Modeling For CO2 AbsorptionDokument10 SeitenRate-Based Modeling For CO2 AbsorptiongoingtohellwithmeNoch keine Bewertungen
- Jonathan Lefebvre, Nike Trudel, Siegfried Bajohr, Thomas KolbDokument9 SeitenJonathan Lefebvre, Nike Trudel, Siegfried Bajohr, Thomas KolbManuel Jose Tabares MontoyaNoch keine Bewertungen
- Absorption With Chemical Reaction. Evaluation of Rate Promoters PDFDokument6 SeitenAbsorption With Chemical Reaction. Evaluation of Rate Promoters PDFmehrdad_k_rNoch keine Bewertungen
- Synthesis and Control Analysis of Gas Absorption Column Using Matlab and SimulinkDokument9 SeitenSynthesis and Control Analysis of Gas Absorption Column Using Matlab and SimulinkLuisana MolinaNoch keine Bewertungen
- 2020 (Plasma) Comparision of Gliding Arc and Micorwave PlasmaDokument25 Seiten2020 (Plasma) Comparision of Gliding Arc and Micorwave PlasmaKin Wai CheahNoch keine Bewertungen
- Co Gasification of Coal and TyreDokument8 SeitenCo Gasification of Coal and TyreJeff Ong Soon HuatNoch keine Bewertungen
- Modeling of Sulphonation of Tridecylbenzene in A Falling Film ReactorDokument13 SeitenModeling of Sulphonation of Tridecylbenzene in A Falling Film Reactoringegnere1234Noch keine Bewertungen
- Simulation of Steam Reformers For MethaneDokument6 SeitenSimulation of Steam Reformers For Methanedashali1100% (1)
- Combustion of Industrial Gas in Porous Media Burner: Hui Liu, Wenzhong Chen Benwen LiDokument4 SeitenCombustion of Industrial Gas in Porous Media Burner: Hui Liu, Wenzhong Chen Benwen LiMuhammad AwaisNoch keine Bewertungen
- Froment1994 PDFDokument14 SeitenFroment1994 PDFDia Fatima MaguikayNoch keine Bewertungen
- Water Gas Shift Reaction Kinetics and Reactor Modeling For Fuel Cell Grade Hydrogen PDFDokument8 SeitenWater Gas Shift Reaction Kinetics and Reactor Modeling For Fuel Cell Grade Hydrogen PDFKmilo BolañosNoch keine Bewertungen
- Intrinsic Kinetics of Dimethyl Ether Synthesis From SyngasDokument7 SeitenIntrinsic Kinetics of Dimethyl Ether Synthesis From SyngasJayeshNoch keine Bewertungen
- AllDokument9 SeitenAllswarhiliNoch keine Bewertungen
- Baur CatTod 2001Dokument8 SeitenBaur CatTod 2001tungksnbNoch keine Bewertungen
- Cfd-Modelling For The Combustion of Solid Baled BiomassDokument6 SeitenCfd-Modelling For The Combustion of Solid Baled BiomassMir Mustafa AliNoch keine Bewertungen
- 0602 Paper Code 602Dokument10 Seiten0602 Paper Code 602Tomás Opazo RodríguezNoch keine Bewertungen
- 2-Mathematical Modeling and Simulation of Hydrotreating Reactors Cocurrent Versus Countercurrent Operations - Art5Dokument14 Seiten2-Mathematical Modeling and Simulation of Hydrotreating Reactors Cocurrent Versus Countercurrent Operations - Art5Vicente SosaNoch keine Bewertungen
- Thermodynamic and Experimental Approach To Ceramic Materials: Gas - Solid/liquid EquilibriaDokument7 SeitenThermodynamic and Experimental Approach To Ceramic Materials: Gas - Solid/liquid Equilibriafofia1955Noch keine Bewertungen
- Gerg 2008 PDFDokument60 SeitenGerg 2008 PDFfddddddNoch keine Bewertungen
- Feng 2005Dokument8 SeitenFeng 2005americo molinaNoch keine Bewertungen
- Chemical Engineering MathematicsDokument103 SeitenChemical Engineering MathematicsRyan NurisalNoch keine Bewertungen
- Simplified Kinetic Models of Methanol Oxidation On PDFDokument18 SeitenSimplified Kinetic Models of Methanol Oxidation On PDFMohammed FaiqNoch keine Bewertungen
- Simplified Kinetic Models of Methanol Oxidation On PDFDokument18 SeitenSimplified Kinetic Models of Methanol Oxidation On PDFMohammed FaiqNoch keine Bewertungen
- 9.1 - Gas-Liquid and Gas-Liquid-Solid ReactionsDokument100 Seiten9.1 - Gas-Liquid and Gas-Liquid-Solid ReactionsHendriyana StNoch keine Bewertungen
- Iupac Intro Het PDFDokument8 SeitenIupac Intro Het PDFRanga SubramanianNoch keine Bewertungen
- Reactivity of Coal Gasification With Steam and CO2Dokument9 SeitenReactivity of Coal Gasification With Steam and CO2udaybhatkandeNoch keine Bewertungen
- 152Dokument9 Seiten152chemsac2Noch keine Bewertungen
- Kinetics and Mechanism of Thermal Decomposition of GaNDokument7 SeitenKinetics and Mechanism of Thermal Decomposition of GaNjhlinjNoch keine Bewertungen
- Dannenberg2000 Article MathematicalModelOfThePEMFCDokument11 SeitenDannenberg2000 Article MathematicalModelOfThePEMFCDEVA NAIKNoch keine Bewertungen
- Gawish SPE Paper PDFDokument11 SeitenGawish SPE Paper PDFEdsonNoch keine Bewertungen
- Non-Isothermal Gas Absorption With Reversible Chemical ReactionDokument16 SeitenNon-Isothermal Gas Absorption With Reversible Chemical Reactiontpqnd90gmailcomNoch keine Bewertungen
- Deutschmann NatGasCS01Dokument8 SeitenDeutschmann NatGasCS01vazzoleralex6884Noch keine Bewertungen
- The Engineering of Hydrogen PeroxideDokument12 SeitenThe Engineering of Hydrogen PeroxideDian Ahmad BudianaNoch keine Bewertungen
- Upaper24 HQTangDokument17 SeitenUpaper24 HQTangSuharman ArmanNoch keine Bewertungen
- Tn287-TN287 Modeling Coal Gasification With CFD and The Discrete Phase MethodDokument9 SeitenTn287-TN287 Modeling Coal Gasification With CFD and The Discrete Phase MethodNenuko CrossNoch keine Bewertungen
- Gas Absorption With Chemical Reaction in Packed PDFDokument5 SeitenGas Absorption With Chemical Reaction in Packed PDFCatherine CcasaNoch keine Bewertungen
- Co2 Captre and Hydrogen Purification From Synthesis Gas by PsaDokument6 SeitenCo2 Captre and Hydrogen Purification From Synthesis Gas by PsaVũ Duy HưngNoch keine Bewertungen
- MechanismWGSonPt Lars 2008Dokument10 SeitenMechanismWGSonPt Lars 2008leonardoNoch keine Bewertungen
- Thermo ChemistryDokument11 SeitenThermo Chemistryamatory1702Noch keine Bewertungen
- Chen 2008Dokument13 SeitenChen 2008AdiSoitNoch keine Bewertungen
- Mass Transfer LaboratoryDokument30 SeitenMass Transfer Laboratoryمحمد عليNoch keine Bewertungen
- Paper Reza SciencedirectDokument14 SeitenPaper Reza Sciencedirectanon_808431206Noch keine Bewertungen
- Tray Efficiency DeterminationDokument10 SeitenTray Efficiency DeterminationtoanNoch keine Bewertungen
- 2004 Methanol Steam Reforming Over CuZnOAl2O3 Catalyst Kinetics and Effectiveness FactorDokument11 Seiten2004 Methanol Steam Reforming Over CuZnOAl2O3 Catalyst Kinetics and Effectiveness FactorChauNoch keine Bewertungen
- Behin Et Al-2013-Chemical Engineering & TechnologyDokument10 SeitenBehin Et Al-2013-Chemical Engineering & TechnologyIgnacio JuanNoch keine Bewertungen
- Simulation of The Catalytic Partial Oxidation of Methane To Synthesis Gas by D.groote, FromentDokument20 SeitenSimulation of The Catalytic Partial Oxidation of Methane To Synthesis Gas by D.groote, FromentMOHAMMAD HASHIM KHAN100% (1)
- InTech-Two Dimensional Pem Fuel Cells Modeling Using Comsol Multiphysics PDFDokument13 SeitenInTech-Two Dimensional Pem Fuel Cells Modeling Using Comsol Multiphysics PDFMostafa AnnakaNoch keine Bewertungen
- 19-2001-Determination of Catalytic Packing Characteristics For Reactive DistillationDokument11 Seiten19-2001-Determination of Catalytic Packing Characteristics For Reactive Distillationehsan zeraatkarNoch keine Bewertungen
- The Partial Oxidation of Methane To Syngas in A Palladium Membrane Reactor: Simulation and Experimental StudiesDokument11 SeitenThe Partial Oxidation of Methane To Syngas in A Palladium Membrane Reactor: Simulation and Experimental StudiesMehul VarshneyNoch keine Bewertungen
- Modeling of CO2 Capture by MEADokument11 SeitenModeling of CO2 Capture by MEAEdison ChoiNoch keine Bewertungen
- CH 204: Chemical Reaction Engineering - Lecture Notes: January-April 2010Dokument56 SeitenCH 204: Chemical Reaction Engineering - Lecture Notes: January-April 2010enjoy your life youh yohNoch keine Bewertungen
- A Modern Course in Statistical PhysicsVon EverandA Modern Course in Statistical PhysicsBewertung: 3.5 von 5 Sternen3.5/5 (2)
- 510 - 2005 Zhang IPE CAS Modeling and Simulation of High Pressure Urea Synthesis LoopDokument10 Seiten510 - 2005 Zhang IPE CAS Modeling and Simulation of High Pressure Urea Synthesis LoopAji PratamaNoch keine Bewertungen
- Mass Transfer (Topic 2)Dokument23 SeitenMass Transfer (Topic 2)Siswand BIn Mohd Ali0% (1)
- Hydrogen Production From Partial Oxidation of Methane Using An AC Rotating Gliding Arc ReactorDokument7 SeitenHydrogen Production From Partial Oxidation of Methane Using An AC Rotating Gliding Arc ReactorMadhu JbNoch keine Bewertungen
- Plug Flow ReactorDokument9 SeitenPlug Flow ReactorTajTajNoch keine Bewertungen
- Electrowinning ZN 1Dokument12 SeitenElectrowinning ZN 1Mhd. Didi Endah PranataNoch keine Bewertungen
- Fuel Cell Workshop For Secondary School Teachers: Fabrication of A Gas Diffusion Electrode Practical SessionDokument6 SeitenFuel Cell Workshop For Secondary School Teachers: Fabrication of A Gas Diffusion Electrode Practical SessionFunsho OlorunyomiNoch keine Bewertungen
- Impact of Carbon Dioxide On Water (A Weak-Strong Challenge) : by Joseph OlorunyomiDokument6 SeitenImpact of Carbon Dioxide On Water (A Weak-Strong Challenge) : by Joseph OlorunyomiFunsho OlorunyomiNoch keine Bewertungen
- Reference ElectrodeDokument1 SeiteReference ElectrodeFunsho Olorunyomi100% (1)
- First CyclesDokument1 SeiteFirst CyclesFunsho OlorunyomiNoch keine Bewertungen
- Financial Intelligence Centre Scams NotificationDokument2 SeitenFinancial Intelligence Centre Scams NotificationFunsho OlorunyomiNoch keine Bewertungen
- Colloid Chemistry PracticeDokument48 SeitenColloid Chemistry PracticeFunsho OlorunyomiNoch keine Bewertungen
- Colloid Chemistry PracticeDokument48 SeitenColloid Chemistry PracticeFunsho OlorunyomiNoch keine Bewertungen
- Hydraulics LaboratoryDokument5 SeitenHydraulics LaboratoryBernard PalmerNoch keine Bewertungen
- SEISMOMETERDokument13 SeitenSEISMOMETERErnesto GullodNoch keine Bewertungen
- NSCP 2015Dokument1.008 SeitenNSCP 2015yanmamalioNoch keine Bewertungen
- Desmi Pumps PDFDokument30 SeitenDesmi Pumps PDFlunatic20000% (1)
- Ijmet 11 07 002Dokument8 SeitenIjmet 11 07 002Siranjeevi AbimanyuNoch keine Bewertungen
- Press Work-Shearing - Bending - Forming - Deep DrawingDokument132 SeitenPress Work-Shearing - Bending - Forming - Deep DrawingAashiq Salu ThekkinedathNoch keine Bewertungen
- Experiment 1: Heat Transfer Measurements Using A Thermal Imaging Infrared CameraDokument6 SeitenExperiment 1: Heat Transfer Measurements Using A Thermal Imaging Infrared CamerapmnitsNoch keine Bewertungen
- STEM - Physics 1 CG - With Tagged Sci EquipmentDokument15 SeitenSTEM - Physics 1 CG - With Tagged Sci EquipmentAndres Kalikasan Sara100% (8)
- Climate Change, Meaning and ImplicationDokument3 SeitenClimate Change, Meaning and ImplicationAbel TaddeleNoch keine Bewertungen
- C-Arm Parts and PrinciplesDokument10 SeitenC-Arm Parts and PrinciplesErshad SohailNoch keine Bewertungen
- Operation and Calibration of UV-VIS SpectrophotometerDokument8 SeitenOperation and Calibration of UV-VIS SpectrophotometerMaruthi K100% (1)
- Level Ii Questions - RTDokument7 SeitenLevel Ii Questions - RTViral ThankiNoch keine Bewertungen
- CBR OptikDokument8 SeitenCBR OptikNuriyaniNoch keine Bewertungen
- Successful PSA of Dry PowdersDokument3 SeitenSuccessful PSA of Dry PowderssiswantoNoch keine Bewertungen
- 2019 2352 AuthorDokument25 Seiten2019 2352 AuthorSharmila BNoch keine Bewertungen
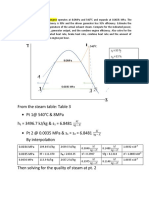
- From The Steam Table: Table 3 PT 1@ 540 C & 8mpa H 3496.7 KJ/KG & S 6.8481 PT 2 at 0.0035 Mpa & S S 6.8481 by InterpolationDokument5 SeitenFrom The Steam Table: Table 3 PT 1@ 540 C & 8mpa H 3496.7 KJ/KG & S 6.8481 PT 2 at 0.0035 Mpa & S S 6.8481 by InterpolationMark AgusNoch keine Bewertungen
- WTP (Water Treatment Plant) DesignDokument3 SeitenWTP (Water Treatment Plant) DesignPratiksha Pawar100% (1)
- Https Eapplication - Nitrkl.ac - in Nitris Student Examination Results Internal Grade Card UG - Aspx Guttr 324432JHGFD67 MTQ2MTI - m9kQIZdicXMDokument2 SeitenHttps Eapplication - Nitrkl.ac - in Nitris Student Examination Results Internal Grade Card UG - Aspx Guttr 324432JHGFD67 MTQ2MTI - m9kQIZdicXMKalinga Prasad BeheraNoch keine Bewertungen
- ScientistDokument2 SeitenScientistJerickson MauricioNoch keine Bewertungen
- Science9 Q4 SLM9Dokument11 SeitenScience9 Q4 SLM9Soliel RiegoNoch keine Bewertungen
- Course Specification: Bachelor of Science in Marine TransportationDokument29 SeitenCourse Specification: Bachelor of Science in Marine TransportationEjay Rich ReglosNoch keine Bewertungen
- MicroscopeDokument4 SeitenMicroscopeBryan PaulNoch keine Bewertungen
- Optical Communications4Dokument19 SeitenOptical Communications4keane1Noch keine Bewertungen
- Heat Geek Mastery Workbook Home Print OffDokument101 SeitenHeat Geek Mastery Workbook Home Print OffJason HaywardNoch keine Bewertungen
- 2021 Uzem Chemistry Ch09 - 2 Solubility and Complex-Ion EquilibriaDokument102 Seiten2021 Uzem Chemistry Ch09 - 2 Solubility and Complex-Ion EquilibriaErdem AltunNoch keine Bewertungen
- 4D Rods 3D Structures Via Programmable 1D Composite R 2018 Materials DesiDokument10 Seiten4D Rods 3D Structures Via Programmable 1D Composite R 2018 Materials DesiJorge Luis Garcia ZuñigaNoch keine Bewertungen
- Letter To Editor On The Effects of Climate Change On The EnvironmentDokument2 SeitenLetter To Editor On The Effects of Climate Change On The EnvironmentKAYANNA LAWRENCENoch keine Bewertungen
- Effect of Turbocharging On Exhaust Brake Performance in An AutomobileDokument6 SeitenEffect of Turbocharging On Exhaust Brake Performance in An Automobileabhisaxena93Noch keine Bewertungen
- Electric Motor Vibration Diagnostic ChartDokument1 SeiteElectric Motor Vibration Diagnostic ChartfazzlieNoch keine Bewertungen
- Notes Chapter 18-1Dokument65 SeitenNotes Chapter 18-1Biruk BtNoch keine Bewertungen