Das könnte Ihnen auch gefallen
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryVon EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryBewertung: 3.5 von 5 Sternen3.5/5 (231)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Von EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Bewertung: 4.5 von 5 Sternen4.5/5 (121)
- Grit: The Power of Passion and PerseveranceVon EverandGrit: The Power of Passion and PerseveranceBewertung: 4 von 5 Sternen4/5 (588)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaVon EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaBewertung: 4.5 von 5 Sternen4.5/5 (266)
- The Little Book of Hygge: Danish Secrets to Happy LivingVon EverandThe Little Book of Hygge: Danish Secrets to Happy LivingBewertung: 3.5 von 5 Sternen3.5/5 (400)
- Never Split the Difference: Negotiating As If Your Life Depended On ItVon EverandNever Split the Difference: Negotiating As If Your Life Depended On ItBewertung: 4.5 von 5 Sternen4.5/5 (838)
- Shoe Dog: A Memoir by the Creator of NikeVon EverandShoe Dog: A Memoir by the Creator of NikeBewertung: 4.5 von 5 Sternen4.5/5 (537)
- The Emperor of All Maladies: A Biography of CancerVon EverandThe Emperor of All Maladies: A Biography of CancerBewertung: 4.5 von 5 Sternen4.5/5 (271)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeVon EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeBewertung: 4 von 5 Sternen4/5 (5794)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyVon EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyBewertung: 3.5 von 5 Sternen3.5/5 (2259)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersVon EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersBewertung: 4.5 von 5 Sternen4.5/5 (344)
- Team of Rivals: The Political Genius of Abraham LincolnVon EverandTeam of Rivals: The Political Genius of Abraham LincolnBewertung: 4.5 von 5 Sternen4.5/5 (234)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreVon EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreBewertung: 4 von 5 Sternen4/5 (1090)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceVon EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceBewertung: 4 von 5 Sternen4/5 (895)
- Her Body and Other Parties: StoriesVon EverandHer Body and Other Parties: StoriesBewertung: 4 von 5 Sternen4/5 (821)
- USMLE Road Map Pharmacology PDFDokument497 SeitenUSMLE Road Map Pharmacology PDFYeshaa Mirani100% (1)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureVon EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureBewertung: 4.5 von 5 Sternen4.5/5 (474)
- The Unwinding: An Inner History of the New AmericaVon EverandThe Unwinding: An Inner History of the New AmericaBewertung: 4 von 5 Sternen4/5 (45)
- The Yellow House: A Memoir (2019 National Book Award Winner)Von EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Bewertung: 4 von 5 Sternen4/5 (98)
- Systemic Response To Injury and Metabolic SupportDokument118 SeitenSystemic Response To Injury and Metabolic SupportNicole DeverasNoch keine Bewertungen
- On Fire: The (Burning) Case for a Green New DealVon EverandOn Fire: The (Burning) Case for a Green New DealBewertung: 4 von 5 Sternen4/5 (73)
- Quiz BiochemistryDokument100 SeitenQuiz BiochemistryMedShare88% (25)
- Biotechnology QuestionsDokument13 SeitenBiotechnology Questionssingamroopa100% (2)
- Pharmaceutical Biotechnology Fundamentals and Applications - Lecture Notes, Study Material and Important Questions, AnswersDokument141 SeitenPharmaceutical Biotechnology Fundamentals and Applications - Lecture Notes, Study Material and Important Questions, AnswersM.V. TV100% (1)
- Metabolism of CarbohydratesDokument26 SeitenMetabolism of CarbohydratesMM Qizill67% (3)
- Pharmacology-I II Pharm.D. Question BankDokument6 SeitenPharmacology-I II Pharm.D. Question BankAnanda Vijayasarathy100% (1)
- Spinal Cord InjuryDokument91 SeitenSpinal Cord InjuryPrabaningrum DwidjoasmoroNoch keine Bewertungen
- Spinal Cord InjuryDokument91 SeitenSpinal Cord InjuryPrabaningrum DwidjoasmoroNoch keine Bewertungen
- TB Drug Induced Liver Injury 27aug2013Dokument39 SeitenTB Drug Induced Liver Injury 27aug2013Edi Uchiha SutarmantoNoch keine Bewertungen
- 6 Enzymes and Cellular Regulation-SDokument5 Seiten6 Enzymes and Cellular Regulation-Sapi-502781581Noch keine Bewertungen
- PPTDokument25 SeitenPPTPrabaningrum DwidjoasmoroNoch keine Bewertungen
- Analgesic Effect of Tms in Chronic Low Back PainDokument9 SeitenAnalgesic Effect of Tms in Chronic Low Back PainPrabaningrum DwidjoasmoroNoch keine Bewertungen
- 3 1 - 13 15Dokument3 Seiten3 1 - 13 15Prabaningrum DwidjoasmoroNoch keine Bewertungen
- Neuroradiology of SpineDokument60 SeitenNeuroradiology of SpinePrabaningrum DwidjoasmoroNoch keine Bewertungen
- Predictive Factors For Postoperative Outcome in Temporal Lobe Epilepsy According To Two Different ClassificationsDokument12 SeitenPredictive Factors For Postoperative Outcome in Temporal Lobe Epilepsy According To Two Different ClassificationsPrabaningrum DwidjoasmoroNoch keine Bewertungen
- Manic Attack Possibly Triggered by SulbuthiamineDokument3 SeitenManic Attack Possibly Triggered by SulbuthiaminePrabaningrum DwidjoasmoroNoch keine Bewertungen
- Anderson 2016Dokument11 SeitenAnderson 2016Prabaningrum DwidjoasmoroNoch keine Bewertungen
- 8.14.07 Tolosa Hunt Syndrome PaglieiDokument36 Seiten8.14.07 Tolosa Hunt Syndrome PaglieiRakasiwi GalihNoch keine Bewertungen
- 10 2 3 29 EDSS - FormDokument2 Seiten10 2 3 29 EDSS - FormChibulcuteanNoch keine Bewertungen
- Stroke-Unit Care For Acute Stroke Patients: An Observational Follow-Up StudyDokument7 SeitenStroke-Unit Care For Acute Stroke Patients: An Observational Follow-Up StudyPrabaningrum DwidjoasmoroNoch keine Bewertungen
- An Update Brain Death CriteriaDokument4 SeitenAn Update Brain Death CriteriaPrabaningrum DwidjoasmoroNoch keine Bewertungen
- Instruction Manual For The ILAE 2017Dokument12 SeitenInstruction Manual For The ILAE 2017Prabaningrum DwidjoasmoroNoch keine Bewertungen
- Hepatotoxicity Related To Anti Tuberculosis DrugsDokument13 SeitenHepatotoxicity Related To Anti Tuberculosis DrugsPrabaningrum DwidjoasmoroNoch keine Bewertungen
- Erb's Palsy Compensation GuideDokument6 SeitenErb's Palsy Compensation GuideMedical Claims OnlineNoch keine Bewertungen
- ILAE Clasiffication and Teminology 2017Dokument10 SeitenILAE Clasiffication and Teminology 2017Prabaningrum DwidjoasmoroNoch keine Bewertungen
- Peripheral Nerve BlockDokument70 SeitenPeripheral Nerve BlockPrabaningrum DwidjoasmoroNoch keine Bewertungen
- Zinc and Cognitive DevelopmentDokument7 SeitenZinc and Cognitive DevelopmentFitriyana WinarnoNoch keine Bewertungen
- DiliDokument18 SeitenDiliKharisma Rizqiah WahyuniNoch keine Bewertungen
- Stroke Unit Duty Report Widya SEPTEMBER 23RD, 2014Dokument18 SeitenStroke Unit Duty Report Widya SEPTEMBER 23RD, 2014Prabaningrum DwidjoasmoroNoch keine Bewertungen
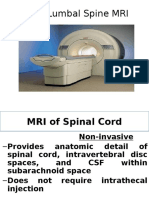
- Widya - Basic Lumbal Spine MriDokument31 SeitenWidya - Basic Lumbal Spine MriPrabaningrum DwidjoasmoroNoch keine Bewertungen
- TransthyretinDokument15 SeitenTransthyretinPrabaningrum DwidjoasmoroNoch keine Bewertungen
- Anesthetic Review LocalDokument37 SeitenAnesthetic Review LocalPrabaningrum Dwidjoasmoro100% (1)
- Lesions of The Spinal CordDokument29 SeitenLesions of The Spinal CordPrabaningrum DwidjoasmoroNoch keine Bewertungen
- Chitinase Inhibitor-LBVSDokument14 SeitenChitinase Inhibitor-LBVSDanyal2222Noch keine Bewertungen
- Mutagenesis PDFDokument8 SeitenMutagenesis PDFGuillermo RomeroNoch keine Bewertungen
- Mark Scheme (Results) Summer 2016: Pearson Edexcel International GCSE in Biology (4BI0) Paper 2BRDokument12 SeitenMark Scheme (Results) Summer 2016: Pearson Edexcel International GCSE in Biology (4BI0) Paper 2BRMostofa ZarifNoch keine Bewertungen
- UntitledDokument339 SeitenUntitledJOS� FRANCISCO G�MEZ RODR�GUEZNoch keine Bewertungen
- Test 1 - APQ Model Answers-1Dokument14 SeitenTest 1 - APQ Model Answers-1Matsiri ImmanuelNoch keine Bewertungen
- PEGFP-N1 Vector InformationDokument3 SeitenPEGFP-N1 Vector InformationNicholas SoNoch keine Bewertungen
- (LMFR) Leanmass Hyper Responders (Cholesterol)Dokument5 Seiten(LMFR) Leanmass Hyper Responders (Cholesterol)simasNoch keine Bewertungen
- Solution of Biology SSC-I (3rd Set)Dokument13 SeitenSolution of Biology SSC-I (3rd Set)Zunaira SafdarNoch keine Bewertungen
- GPAT Orientation PDFDokument28 SeitenGPAT Orientation PDFDrGajanan Vaishnav33% (3)
- Calculation Examples Feeding Values For RuminantsDokument8 SeitenCalculation Examples Feeding Values For RuminantsВолодимир Сидоренко100% (1)
- BS-800M BS-800M: Clinical Chemistry Solution Clinical Chemistry SolutionDokument4 SeitenBS-800M BS-800M: Clinical Chemistry Solution Clinical Chemistry Solutionads241167Noch keine Bewertungen
- TMP D820Dokument9 SeitenTMP D820FrontiersNoch keine Bewertungen
- Anaphy Lab - Activity in Cell and MicroscopeDokument5 SeitenAnaphy Lab - Activity in Cell and MicroscopeAlvin Cris RongavillaNoch keine Bewertungen
- 2019 PDFDokument15 Seiten2019 PDFneculavNoch keine Bewertungen
- This Study Resource Was: DNA Base Pairing WorksheetDokument5 SeitenThis Study Resource Was: DNA Base Pairing Worksheetphil tolentinoNoch keine Bewertungen
- Answers: Biology 9744/03Dokument8 SeitenAnswers: Biology 9744/03Timothy HandokoNoch keine Bewertungen
- 1.3 WorksheetDokument7 Seiten1.3 WorksheetFRENCHONLYNoch keine Bewertungen
- Summative Test in MacromoleculesDokument3 SeitenSummative Test in MacromoleculesCalebamaziah BonzonNoch keine Bewertungen
- 13-14 MBB306 Lecture 5 The Herpesviruses - RJDokument28 Seiten13-14 MBB306 Lecture 5 The Herpesviruses - RJAnonymous PyUR1NINoch keine Bewertungen
- ArenavirusDokument29 SeitenArenavirusRamirez GiovarNoch keine Bewertungen
- Bacterialand Archael InclusionsDokument15 SeitenBacterialand Archael InclusionsXimena AlcántaraNoch keine Bewertungen
- Isolation of Plant Genomic Dna by Ctab MethodDokument4 SeitenIsolation of Plant Genomic Dna by Ctab MethodVikram ArjunNoch keine Bewertungen