Das könnte Ihnen auch gefallen
- 11 - Chapter 5Dokument19 Seiten11 - Chapter 5The FrequencyNoch keine Bewertungen
- Abnormal Spermatogenesis and Reduced Fertility in Transition Nuclear Protein 1-Deficient MiceDokument6 SeitenAbnormal Spermatogenesis and Reduced Fertility in Transition Nuclear Protein 1-Deficient MiceBerlinNoch keine Bewertungen
- Teratozoospermia in Mice Lacking The Transition Protein 2 (Tnp2)Dokument8 SeitenTeratozoospermia in Mice Lacking The Transition Protein 2 (Tnp2)BerlinNoch keine Bewertungen
- Maki Murakami 1997 Formation of Potent Hybrid Promoters of The Mutant LLM Gene by Is256 Transposition in MethicillinDokument5 SeitenMaki Murakami 1997 Formation of Potent Hybrid Promoters of The Mutant LLM Gene by Is256 Transposition in MethicillingaryNoch keine Bewertungen
- A Global Increase in 5-Hydroxymethylcytosine Levels Marks Osteoarthritic ChondrocytesDokument11 SeitenA Global Increase in 5-Hydroxymethylcytosine Levels Marks Osteoarthritic Chondrocytescriters007Noch keine Bewertungen
- Escherichia ColiDokument10 SeitenEscherichia ColisurendrasrawanNoch keine Bewertungen
- PTEN Draft2Dokument10 SeitenPTEN Draft2jenna_bugabooNoch keine Bewertungen
- Human Indolethylamine N-Methyltransferase: cDNA Cloning and Expression, Gene Cloning, and Chromosomal LocalizationDokument13 SeitenHuman Indolethylamine N-Methyltransferase: cDNA Cloning and Expression, Gene Cloning, and Chromosomal LocalizationnndimethylNoch keine Bewertungen
- A Semi Synthetic Organism II Carolina Nature 2017Dokument20 SeitenA Semi Synthetic Organism II Carolina Nature 2017José Antonio Silva NetoNoch keine Bewertungen
- DNA Repair Protein Involved in Heart and Blood DevelopmentDokument12 SeitenDNA Repair Protein Involved in Heart and Blood DevelopmentSol Jumaide WerbleNoch keine Bewertungen
- Español Estrogen Down Regulates COMT Transcription Via Promoter DNA Methylation in Human Breast Cancer Cells Sin ImagenesDokument16 SeitenEspañol Estrogen Down Regulates COMT Transcription Via Promoter DNA Methylation in Human Breast Cancer Cells Sin ImagenesJessica Becerra CarocaNoch keine Bewertungen
- TFG3/TAF30/ANC1, A Component of The Yeast SWI/SNF Complex That Is Similar To The Leukemogenic Proteins ENL and AF-9Dokument9 SeitenTFG3/TAF30/ANC1, A Component of The Yeast SWI/SNF Complex That Is Similar To The Leukemogenic Proteins ENL and AF-9Eva Benito BerrocalNoch keine Bewertungen
- Spermatozoa NDokument5 SeitenSpermatozoa Narulsidd74Noch keine Bewertungen
- Allozyme Variations of Trichoderma Harzianum and Its Taxonomic ImplicationsDokument8 SeitenAllozyme Variations of Trichoderma Harzianum and Its Taxonomic Implicationsray m deraniaNoch keine Bewertungen
- Expression of Human Soluble Tumor Necrosis Factor (TNF) - Related Apoptosis-Inducing Ligand in Transplastomic TobaccoDokument8 SeitenExpression of Human Soluble Tumor Necrosis Factor (TNF) - Related Apoptosis-Inducing Ligand in Transplastomic TobaccoasgharfeiziNoch keine Bewertungen
- PubmedDokument7 SeitenPubmedcristinaNoch keine Bewertungen
- Characterization of Tryptophan Aminotransferase 1 of Malassezia Furfur, The Key Enzyme in The Production of Indolic Compounds by M. FurfurDokument6 SeitenCharacterization of Tryptophan Aminotransferase 1 of Malassezia Furfur, The Key Enzyme in The Production of Indolic Compounds by M. FurfurMonica AntolinezNoch keine Bewertungen
- Artigo ThiagoDokument6 SeitenArtigo ThiagoFelippe MousovichNoch keine Bewertungen
- Differential Display RT PCR of Total RNA From Human Foreskin Fibroblasts For Investigation of Androgen-Dependent Gene ExpressionDokument8 SeitenDifferential Display RT PCR of Total RNA From Human Foreskin Fibroblasts For Investigation of Androgen-Dependent Gene ExpressionAltaicaNoch keine Bewertungen
- Crosstalk Between Normal and Tumoral BraDokument7 SeitenCrosstalk Between Normal and Tumoral Bravictorubong404Noch keine Bewertungen
- Gsh1 Pgpa Leishmania TarentolaeDokument9 SeitenGsh1 Pgpa Leishmania TarentolaeVamsi Krishna ThiriveedhiNoch keine Bewertungen
- Telomerase Reverse Transcriptase mRNA Expression and Telomerase Activity in Hepatocellular CarcinomaDokument5 SeitenTelomerase Reverse Transcriptase mRNA Expression and Telomerase Activity in Hepatocellular CarcinomaSyief Schareez ZlightNoch keine Bewertungen
- Utilizado - JoenjeDokument4 SeitenUtilizado - JoenjedeliveryworkNoch keine Bewertungen
- Characterization of tRNA Precursor Splicing in Mammalian ExtractsDokument7 SeitenCharacterization of tRNA Precursor Splicing in Mammalian ExtractsfrostyNoch keine Bewertungen
- Denecke HumMutat 06Dokument8 SeitenDenecke HumMutat 06Thorsten MarquardtNoch keine Bewertungen
- Association of Tnfa, Tnfr1, and Tnfr2 Polymorphisms With Sperm Concentration and MotilityDokument7 SeitenAssociation of Tnfa, Tnfr1, and Tnfr2 Polymorphisms With Sperm Concentration and MotilityInneke NoerNoch keine Bewertungen
- INVESTIGACIÓNDokument8 SeitenINVESTIGACIÓNLuis Rodrigo Alagón PalominoNoch keine Bewertungen
- tmp83FE TMPDokument11 Seitentmp83FE TMPFrontiersNoch keine Bewertungen
- BypassDokument5 SeitenBypassGeorgina HernandezNoch keine Bewertungen
- Geranylgeraniol Is A Potent Inducer of Apoptosis in Tumor CellsDokument3 SeitenGeranylgeraniol Is A Potent Inducer of Apoptosis in Tumor CellsmanumangalNoch keine Bewertungen
- 2686 PDFDokument11 Seiten2686 PDFmojNoch keine Bewertungen
- Ostoa-Saloma Et Al-1997-European Journal of Biochemistry PDFDokument6 SeitenOstoa-Saloma Et Al-1997-European Journal of Biochemistry PDFErik TeránNoch keine Bewertungen
- Chromosomal Aberrations Induced by 5-Azacytidine Combined With VP-16 (Etoposide) in CHO-K1 and XRS-5 Cell LinesDokument16 SeitenChromosomal Aberrations Induced by 5-Azacytidine Combined With VP-16 (Etoposide) in CHO-K1 and XRS-5 Cell LinesAry AguiarNoch keine Bewertungen
- Duan 2019Dokument4 SeitenDuan 2019Srinath ReddyNoch keine Bewertungen
- Good PaperDokument12 SeitenGood Paperlkanth1Noch keine Bewertungen
- CH 1Dokument3 SeitenCH 1Aqsa ParveenNoch keine Bewertungen
- Plant Selectable Marker Luciferase Gene in Plant CellsDokument4 SeitenPlant Selectable Marker Luciferase Gene in Plant CellsMd Mostafa Kamal AbirNoch keine Bewertungen
- Science 1997Dokument3 SeitenScience 1997demmie1989Noch keine Bewertungen
- Pi Is 0092867400808351Dokument10 SeitenPi Is 0092867400808351MARIA ANGGIE CANTIKA DEWANINoch keine Bewertungen
- Nar00064 0196Dokument8 SeitenNar00064 0196Pipe pelaezNoch keine Bewertungen
- Deletions Affecting The Transposition of An Antibiotic Resistance GeneDokument5 SeitenDeletions Affecting The Transposition of An Antibiotic Resistance GeneChris Letchford-JonesNoch keine Bewertungen
- CYP06 - (Paper) 2004 MurayamaDokument7 SeitenCYP06 - (Paper) 2004 MurayamaCindy ChanNoch keine Bewertungen
- Purification and of Type-I Topoisomerase From Cultured Tobacco Cells1Dokument8 SeitenPurification and of Type-I Topoisomerase From Cultured Tobacco Cells1Abhishek KanyalNoch keine Bewertungen
- AssstDokument5 SeitenAssstDiegoNoch keine Bewertungen
- Degraaff 1996Dokument7 SeitenDegraaff 1996Araceli Enríquez OvandoNoch keine Bewertungen
- PMA Up-Regulates The Transcription of Axl by AP-1 Transcription Factor Binding To TRE Sequences Via The MAPK Cascade in Leukaemia CellsDokument19 SeitenPMA Up-Regulates The Transcription of Axl by AP-1 Transcription Factor Binding To TRE Sequences Via The MAPK Cascade in Leukaemia CellsWilliam RamirezNoch keine Bewertungen
- Mutagenesis High-Affinity: Site-Directed Streptavidin-Biotin Complex: Contributions Tryptophan Residues 79, 108Dokument5 SeitenMutagenesis High-Affinity: Site-Directed Streptavidin-Biotin Complex: Contributions Tryptophan Residues 79, 108Guhan KANoch keine Bewertungen
- Apoptosis 01Dokument10 SeitenApoptosis 01Kamila Środa-PomianekNoch keine Bewertungen
- Membrane-Bound Serine Protease Matriptase-2 (Tmprss6) Is An Essential Regulator of Iron HomeostasisDokument8 SeitenMembrane-Bound Serine Protease Matriptase-2 (Tmprss6) Is An Essential Regulator of Iron HomeostasisAniki PuspitaNoch keine Bewertungen
- Target Cells For Gene TransformationDokument30 SeitenTarget Cells For Gene TransformationnavkirNoch keine Bewertungen
- pRS415Dokument12 SeitenpRS415DiegoNoch keine Bewertungen
- Paper 3Dokument5 SeitenPaper 3AntoniaNoch keine Bewertungen
- Novel Family of Sensor Histidine Kinase Genes in ADokument6 SeitenNovel Family of Sensor Histidine Kinase Genes in AFrancisco EspinosaNoch keine Bewertungen
- Flow CytometryDokument10 SeitenFlow CytometryDaniel BarrigaNoch keine Bewertungen
- 1996 - Determination of Life-Span in Caenorhabditis Elegans by Four Clock GenesDokument5 Seiten1996 - Determination of Life-Span in Caenorhabditis Elegans by Four Clock GenesRaymond LaBoyNoch keine Bewertungen
- Significance of The Patched Gene in Developmental Biology: Evan Zhou T05 CMMB 403Dokument9 SeitenSignificance of The Patched Gene in Developmental Biology: Evan Zhou T05 CMMB 403EvanNoch keine Bewertungen
- Biophysical Characteristics The Replication Arrest ProteinDokument8 SeitenBiophysical Characteristics The Replication Arrest Proteinrfahad22926Noch keine Bewertungen
- Pic RenderDokument9 SeitenPic Renderkora_c995Noch keine Bewertungen
- Environmental Mutagen Society of Japan 19th AnnualDokument2 SeitenEnvironmental Mutagen Society of Japan 19th AnnualQ-MT Branch HQ 497 OM&MNoch keine Bewertungen
- 1130 ACLS HeidenreichDokument43 Seiten1130 ACLS Heidenreichdydy_7193Noch keine Bewertungen
- SumberDokument1 SeiteSumberKalista ApriyaniNoch keine Bewertungen
- ACLS Cardiac Arrest AlgorithmDokument1 SeiteACLS Cardiac Arrest AlgorithmKalista ApriyaniNoch keine Bewertungen
- Acute Urinary Retention: Ronald TanggoDokument63 SeitenAcute Urinary Retention: Ronald TanggoKalista ApriyaniNoch keine Bewertungen
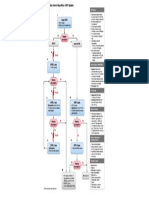
- Diagnosis and Management For Urosepsis PDFDokument9 SeitenDiagnosis and Management For Urosepsis PDFLeon L GayaNoch keine Bewertungen
- If ConditionalDokument7 SeitenIf ConditionalKalista ApriyaniNoch keine Bewertungen
- Worksheet Plant Cell and OsmosisDokument4 SeitenWorksheet Plant Cell and OsmosisKalista ApriyaniNoch keine Bewertungen
- Molecular Basis of Tyrosinase-Negative Oculocutaneous AlbinismDokument7 SeitenMolecular Basis of Tyrosinase-Negative Oculocutaneous AlbinismKalista ApriyaniNoch keine Bewertungen
- Angel Number 1208 Meaning Increased FaithDokument1 SeiteAngel Number 1208 Meaning Increased FaithKhally KatieNoch keine Bewertungen
- TLS FinalDokument69 SeitenTLS FinalGrace Arthur100% (1)
- 1990 Pekoz Design of Cold-Formed Steel Screw ConnectionsDokument15 Seiten1990 Pekoz Design of Cold-Formed Steel Screw ConnectionsmabuhamdNoch keine Bewertungen
- Baltimore Catechism No. 2 (Of 4)Dokument64 SeitenBaltimore Catechism No. 2 (Of 4)gogelNoch keine Bewertungen
- Tamil Ilakkanam Books For TNPSCDokument113 SeitenTamil Ilakkanam Books For TNPSCkk_kamalakkannan100% (1)
- Effects of Alcohol, Tobacco, and Marijuana - PR 1Dokument11 SeitenEffects of Alcohol, Tobacco, and Marijuana - PR 1Mark Andris GempisawNoch keine Bewertungen
- Full Download Social Animal 14th Edition Aronson Test BankDokument35 SeitenFull Download Social Animal 14th Edition Aronson Test Banknaeensiyev100% (32)
- Sample Internship PPTDokument19 SeitenSample Internship PPTSangeeta JamadarNoch keine Bewertungen
- International Conference On Basic Science (ICBS)Dokument22 SeitenInternational Conference On Basic Science (ICBS)repositoryIPBNoch keine Bewertungen
- Nin/Pmjay Id Name of The Vaccination Site Category Type District BlockDokument2 SeitenNin/Pmjay Id Name of The Vaccination Site Category Type District BlockNikunja PadhanNoch keine Bewertungen
- (Durt, - Christoph - Fuchs, - Thomas - Tewes, - Christian) Embodiment, Enaction, and Culture PDFDokument451 Seiten(Durt, - Christoph - Fuchs, - Thomas - Tewes, - Christian) Embodiment, Enaction, and Culture PDFnlf2205100% (3)
- Middle Grades ReportDokument138 SeitenMiddle Grades ReportcraignewmanNoch keine Bewertungen
- Diagnosis: Acute GastroenteritisDokument1 SeiteDiagnosis: Acute GastroenteritisSakshi RanabhatNoch keine Bewertungen
- Gigold PDFDokument61 SeitenGigold PDFSurender SinghNoch keine Bewertungen
- English Lesson PlanDokument3 SeitenEnglish Lesson PlanJeremias MartirezNoch keine Bewertungen
- 50 p7 Kids AvikdeDokument2 Seiten50 p7 Kids AvikdebankansNoch keine Bewertungen
- Moon and SaturnDokument4 SeitenMoon and SaturnRamanasarmaNoch keine Bewertungen
- BSCHMCTT 101Dokument308 SeitenBSCHMCTT 101JITTUNoch keine Bewertungen
- Second Grading EappDokument2 SeitenSecond Grading EappConnieRoseRamos100% (2)
- Nguyễn Thị Ngọc Huyền - 19125516 - Homework 3Dokument7 SeitenNguyễn Thị Ngọc Huyền - 19125516 - Homework 3Nguyễn HuyềnNoch keine Bewertungen
- This Study Resource Was: Interactive Reading QuestionsDokument3 SeitenThis Study Resource Was: Interactive Reading QuestionsJoshua LagonoyNoch keine Bewertungen
- Boden 2015 Mass Media Playground of StereotypingDokument16 SeitenBoden 2015 Mass Media Playground of StereotypingMiguel CuevaNoch keine Bewertungen
- Lesson I. Background InformationDokument21 SeitenLesson I. Background InformationsuidivoNoch keine Bewertungen
- Ross, D. (2013) - Field Guide To Jumping Spiders of Southeast Idaho.Dokument4 SeitenRoss, D. (2013) - Field Guide To Jumping Spiders of Southeast Idaho.Dave RossNoch keine Bewertungen
- Chan Sophia ResumeDokument1 SeiteChan Sophia Resumeapi-568119902Noch keine Bewertungen
- Attery: User Guide Dict Release 2020Dokument47 SeitenAttery: User Guide Dict Release 2020diegoNoch keine Bewertungen
- Congental Abdominal Wall DefectsDokument38 SeitenCongental Abdominal Wall DefectsAhmad Abu KushNoch keine Bewertungen
- Financial Crisis Among UTHM StudentsDokument7 SeitenFinancial Crisis Among UTHM StudentsPravin PeriasamyNoch keine Bewertungen
- RFP On Internal AuditDokument33 SeitenRFP On Internal AuditCan dien tu Thai Binh DuongNoch keine Bewertungen
- Who, Summary NotesDokument12 SeitenWho, Summary NotesIvan Lohr100% (2)
- Why We Die: The New Science of Aging and the Quest for ImmortalityVon EverandWhy We Die: The New Science of Aging and the Quest for ImmortalityBewertung: 4 von 5 Sternen4/5 (3)
- When the Body Says No by Gabor Maté: Key Takeaways, Summary & AnalysisVon EverandWhen the Body Says No by Gabor Maté: Key Takeaways, Summary & AnalysisBewertung: 3.5 von 5 Sternen3.5/5 (2)
- Gut: the new and revised Sunday Times bestsellerVon EverandGut: the new and revised Sunday Times bestsellerBewertung: 4 von 5 Sternen4/5 (392)
- All That Remains: A Renowned Forensic Scientist on Death, Mortality, and Solving CrimesVon EverandAll That Remains: A Renowned Forensic Scientist on Death, Mortality, and Solving CrimesBewertung: 4.5 von 5 Sternen4.5/5 (397)
- Masterminds: Genius, DNA, and the Quest to Rewrite LifeVon EverandMasterminds: Genius, DNA, and the Quest to Rewrite LifeNoch keine Bewertungen
- A Brief History of Intelligence: Evolution, AI, and the Five Breakthroughs That Made Our BrainsVon EverandA Brief History of Intelligence: Evolution, AI, and the Five Breakthroughs That Made Our BrainsBewertung: 4.5 von 5 Sternen4.5/5 (6)
- The Molecule of More: How a Single Chemical in Your Brain Drives Love, Sex, and Creativity--and Will Determine the Fate of the Human RaceVon EverandThe Molecule of More: How a Single Chemical in Your Brain Drives Love, Sex, and Creativity--and Will Determine the Fate of the Human RaceBewertung: 4.5 von 5 Sternen4.5/5 (516)
- Gut: The Inside Story of Our Body's Most Underrated Organ (Revised Edition)Von EverandGut: The Inside Story of Our Body's Most Underrated Organ (Revised Edition)Bewertung: 4 von 5 Sternen4/5 (378)
- 10% Human: How Your Body's Microbes Hold the Key to Health and HappinessVon Everand10% Human: How Your Body's Microbes Hold the Key to Health and HappinessBewertung: 4 von 5 Sternen4/5 (33)
- Tales from Both Sides of the Brain: A Life in NeuroscienceVon EverandTales from Both Sides of the Brain: A Life in NeuroscienceBewertung: 3 von 5 Sternen3/5 (18)
- The Ancestor's Tale: A Pilgrimage to the Dawn of EvolutionVon EverandThe Ancestor's Tale: A Pilgrimage to the Dawn of EvolutionBewertung: 4 von 5 Sternen4/5 (812)
- Fast Asleep: Improve Brain Function, Lose Weight, Boost Your Mood, Reduce Stress, and Become a Better SleeperVon EverandFast Asleep: Improve Brain Function, Lose Weight, Boost Your Mood, Reduce Stress, and Become a Better SleeperBewertung: 4.5 von 5 Sternen4.5/5 (15)
- Good Without God: What a Billion Nonreligious People Do BelieveVon EverandGood Without God: What a Billion Nonreligious People Do BelieveBewertung: 4 von 5 Sternen4/5 (66)
- A Series of Fortunate Events: Chance and the Making of the Planet, Life, and YouVon EverandA Series of Fortunate Events: Chance and the Making of the Planet, Life, and YouBewertung: 4.5 von 5 Sternen4.5/5 (62)
- Seven and a Half Lessons About the BrainVon EverandSeven and a Half Lessons About the BrainBewertung: 4 von 5 Sternen4/5 (109)
- Undeniable: How Biology Confirms Our Intuition That Life Is DesignedVon EverandUndeniable: How Biology Confirms Our Intuition That Life Is DesignedBewertung: 4 von 5 Sternen4/5 (11)
- Who's in Charge?: Free Will and the Science of the BrainVon EverandWho's in Charge?: Free Will and the Science of the BrainBewertung: 4 von 5 Sternen4/5 (65)
- The Other Side of Normal: How Biology Is Providing the Clues to Unlock the Secrets of Normal and Abnormal BehaviorVon EverandThe Other Side of Normal: How Biology Is Providing the Clues to Unlock the Secrets of Normal and Abnormal BehaviorNoch keine Bewertungen
- Human: The Science Behind What Makes Your Brain UniqueVon EverandHuman: The Science Behind What Makes Your Brain UniqueBewertung: 3.5 von 5 Sternen3.5/5 (38)
- The Rise and Fall of the Dinosaurs: A New History of a Lost WorldVon EverandThe Rise and Fall of the Dinosaurs: A New History of a Lost WorldBewertung: 4 von 5 Sternen4/5 (595)
- The Consciousness Instinct: Unraveling the Mystery of How the Brain Makes the MindVon EverandThe Consciousness Instinct: Unraveling the Mystery of How the Brain Makes the MindBewertung: 4.5 von 5 Sternen4.5/5 (93)
- Moral Tribes: Emotion, Reason, and the Gap Between Us and ThemVon EverandMoral Tribes: Emotion, Reason, and the Gap Between Us and ThemBewertung: 4.5 von 5 Sternen4.5/5 (115)
- Wayfinding: The Science and Mystery of How Humans Navigate the WorldVon EverandWayfinding: The Science and Mystery of How Humans Navigate the WorldBewertung: 4.5 von 5 Sternen4.5/5 (18)
- Why We Sleep: Unlocking the Power of Sleep and DreamsVon EverandWhy We Sleep: Unlocking the Power of Sleep and DreamsBewertung: 4.5 von 5 Sternen4.5/5 (2083)
- The Second Brain: A Groundbreaking New Understanding of Nervous Disorders of the Stomach and IntestineVon EverandThe Second Brain: A Groundbreaking New Understanding of Nervous Disorders of the Stomach and IntestineBewertung: 4 von 5 Sternen4/5 (17)
- Buddha's Brain: The Practical Neuroscience of Happiness, Love & WisdomVon EverandBuddha's Brain: The Practical Neuroscience of Happiness, Love & WisdomBewertung: 4 von 5 Sternen4/5 (215)