Das könnte Ihnen auch gefallen
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Von EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Bewertung: 4.5 von 5 Sternen4.5/5 (119)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaVon EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaBewertung: 4.5 von 5 Sternen4.5/5 (265)
- The Little Book of Hygge: Danish Secrets to Happy LivingVon EverandThe Little Book of Hygge: Danish Secrets to Happy LivingBewertung: 3.5 von 5 Sternen3.5/5 (399)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryVon EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryBewertung: 3.5 von 5 Sternen3.5/5 (231)
- Grit: The Power of Passion and PerseveranceVon EverandGrit: The Power of Passion and PerseveranceBewertung: 4 von 5 Sternen4/5 (587)
- Never Split the Difference: Negotiating As If Your Life Depended On ItVon EverandNever Split the Difference: Negotiating As If Your Life Depended On ItBewertung: 4.5 von 5 Sternen4.5/5 (838)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeVon EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeBewertung: 4 von 5 Sternen4/5 (5794)
- Team of Rivals: The Political Genius of Abraham LincolnVon EverandTeam of Rivals: The Political Genius of Abraham LincolnBewertung: 4.5 von 5 Sternen4.5/5 (234)
- Shoe Dog: A Memoir by the Creator of NikeVon EverandShoe Dog: A Memoir by the Creator of NikeBewertung: 4.5 von 5 Sternen4.5/5 (537)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyVon EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyBewertung: 3.5 von 5 Sternen3.5/5 (2219)
- The Emperor of All Maladies: A Biography of CancerVon EverandThe Emperor of All Maladies: A Biography of CancerBewertung: 4.5 von 5 Sternen4.5/5 (271)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreVon EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreBewertung: 4 von 5 Sternen4/5 (1090)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersVon EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersBewertung: 4.5 von 5 Sternen4.5/5 (344)
- Her Body and Other Parties: StoriesVon EverandHer Body and Other Parties: StoriesBewertung: 4 von 5 Sternen4/5 (821)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceVon EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceBewertung: 4 von 5 Sternen4/5 (894)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureVon EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureBewertung: 4.5 von 5 Sternen4.5/5 (474)
- Lab 3 - Intro To DynamicDokument36 SeitenLab 3 - Intro To DynamicRacheal KirbyNoch keine Bewertungen
- The Unwinding: An Inner History of the New AmericaVon EverandThe Unwinding: An Inner History of the New AmericaBewertung: 4 von 5 Sternen4/5 (45)
- The Yellow House: A Memoir (2019 National Book Award Winner)Von EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Bewertung: 4 von 5 Sternen4/5 (98)
- On Fire: The (Burning) Case for a Green New DealVon EverandOn Fire: The (Burning) Case for a Green New DealBewertung: 4 von 5 Sternen4/5 (73)
- The Theory of Engineering DrawingDokument370 SeitenThe Theory of Engineering Drawingcocotess100% (1)
- Zeta PotentialDokument8 SeitenZeta Potentialapi-3721576100% (2)
- Chemical BondingDokument6 SeitenChemical BondingNoongju AbdullahNoch keine Bewertungen
- GE Gas Turbine IGV AngleDokument10 SeitenGE Gas Turbine IGV AngleSamir BenabdallahNoch keine Bewertungen
- Electromagnetic SpectrumDokument74 SeitenElectromagnetic SpectrumGuiller Lanuza100% (1)
- Alginate Wound DressingsDokument5 SeitenAlginate Wound Dressingssins1984Noch keine Bewertungen
- Are Swedes Really That Normal?Dokument4 SeitenAre Swedes Really That Normal?sins1984Noch keine Bewertungen
- The Design of Prosperity 2009Dokument15 SeitenThe Design of Prosperity 2009sins1984Noch keine Bewertungen
- Chitosan in Bio MaterialDokument22 SeitenChitosan in Bio Materialsins1984Noch keine Bewertungen
- Basic Sensors and Sensing PrinciplesDokument18 SeitenBasic Sensors and Sensing Principlessins1984Noch keine Bewertungen
- High Tech Firefighters ClothingDokument7 SeitenHigh Tech Firefighters Clothingsins1984Noch keine Bewertungen
- PEDOT SD ArticleDokument3 SeitenPEDOT SD Articlesins1984Noch keine Bewertungen
- Improved Dispersion of Carbon Nanotubes in Chitosan - FulltextDokument6 SeitenImproved Dispersion of Carbon Nanotubes in Chitosan - Fulltextsins1984Noch keine Bewertungen
- Clothing Physiology of Surgery Room: Ubaid Ur Rehman Eusuf ZaiDokument8 SeitenClothing Physiology of Surgery Room: Ubaid Ur Rehman Eusuf Zaisins1984Noch keine Bewertungen
- Effect of Hydroxyethylation On Structure and Properties of Chitosan Fibers - 30024433Dokument10 SeitenEffect of Hydroxyethylation On Structure and Properties of Chitosan Fibers - 30024433sins1984Noch keine Bewertungen
- A Review of Chitin and Chitosan ApplicationsDokument27 SeitenA Review of Chitin and Chitosan Applicationssins1984100% (2)
- SOP - UbaidDokument1 SeiteSOP - Ubaidsins1984Noch keine Bewertungen
- Chitosan - Crawling From Crab Shells To Bio Textiles 02Dokument5 SeitenChitosan - Crawling From Crab Shells To Bio Textiles 02sins1984Noch keine Bewertungen
- Invest-Establish in Sjuharad - IsADokument3 SeitenInvest-Establish in Sjuharad - IsAsins1984Noch keine Bewertungen
- Principle Alginate DressingsDokument9 SeitenPrinciple Alginate Dressingssins1984Noch keine Bewertungen
- An Open Issue For Electronic TextilesDokument10 SeitenAn Open Issue For Electronic Textilessins1984Noch keine Bewertungen
- Amicor - Acrylics Antimicrobial FiberDokument10 SeitenAmicor - Acrylics Antimicrobial Fibersins1984Noch keine Bewertungen
- Earth QuizDokument15 SeitenEarth Quizsins1984Noch keine Bewertungen
- Alginates QuestionsDokument7 SeitenAlginates Questionssins1984Noch keine Bewertungen
- Project Proposal For For Masters Thesis in Wound Care ProductsDokument3 SeitenProject Proposal For For Masters Thesis in Wound Care Productssins1984100% (2)
- Textile Composite Wound DressingDokument4 SeitenTextile Composite Wound Dressingsins1984Noch keine Bewertungen
- Basic Human Pathology - Lecture 6Dokument40 SeitenBasic Human Pathology - Lecture 6sins1984Noch keine Bewertungen
- Wound Management Training Holden 07Dokument14 SeitenWound Management Training Holden 07sins1984Noch keine Bewertungen
- Wound Healing Acceleration by A Textile Dressing Containing DBCH and ChitinDokument6 SeitenWound Healing Acceleration by A Textile Dressing Containing DBCH and Chitinsins1984100% (1)
- Modern Wound Care - Lohmann & RauscherDokument18 SeitenModern Wound Care - Lohmann & Rauschersins1984Noch keine Bewertungen
- Tissue Engineering, Drug Delivery, Wound Healing Via Polymer Nano Fibers and NanotubesDokument28 SeitenTissue Engineering, Drug Delivery, Wound Healing Via Polymer Nano Fibers and Nanotubessins1984Noch keine Bewertungen
- Basic Human Pathology - Lecture 5Dokument36 SeitenBasic Human Pathology - Lecture 5sins1984Noch keine Bewertungen
- TCL Air Conditioner Service ManualDokument138 SeitenTCL Air Conditioner Service ManualFabian EtcheniqueNoch keine Bewertungen
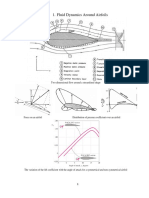
- Overview Aerodynamics 2017Dokument10 SeitenOverview Aerodynamics 2017marcoNoch keine Bewertungen
- Electronics Engg.: Detailed Solutions ofDokument52 SeitenElectronics Engg.: Detailed Solutions ofAshish ChoudharyNoch keine Bewertungen
- Midas FEADokument2 SeitenMidas FEACristian Camilo Londoño PiedrahítaNoch keine Bewertungen
- Compressive Strength of Hydraulic Cement Mortars (Using 2-In. or (50-mm) Cube Specimens)Dokument9 SeitenCompressive Strength of Hydraulic Cement Mortars (Using 2-In. or (50-mm) Cube Specimens)Jesús Luis Arce GuillermoNoch keine Bewertungen
- Irjet V5i5256 PDFDokument5 SeitenIrjet V5i5256 PDFMuhsinaNoch keine Bewertungen
- Electrical and Optical Properties of Indium-Tin Oxide (ITO) Films by Ion-Assisted Deposition (IAD) at Room TemperatureDokument6 SeitenElectrical and Optical Properties of Indium-Tin Oxide (ITO) Films by Ion-Assisted Deposition (IAD) at Room Temperaturereza mirzakhaniNoch keine Bewertungen
- The Mode of Eruptions and Their Tephra Deposits: Tetsuo K and Mitsuru ODokument8 SeitenThe Mode of Eruptions and Their Tephra Deposits: Tetsuo K and Mitsuru OAnggit Tri AtmajaNoch keine Bewertungen
- Activity MergedDokument9 SeitenActivity MergedSoham MondalNoch keine Bewertungen
- Marsh FunnelDokument2 SeitenMarsh Funnel123shripadNoch keine Bewertungen
- Daftar PustakaDokument5 SeitenDaftar PustakamaisyaraaaahNoch keine Bewertungen
- Astm F1717-21Dokument11 SeitenAstm F1717-21wenhsiaochuanNoch keine Bewertungen
- De Electric Circuits EeDokument16 SeitenDe Electric Circuits EeLilet P. DalisayNoch keine Bewertungen
- Mid-Term Math Exam for Grade 5Dokument18 SeitenMid-Term Math Exam for Grade 5李安逸Noch keine Bewertungen
- EagleBurgmann Statotherm P Foil 9591 P enDokument1 SeiteEagleBurgmann Statotherm P Foil 9591 P enkeyur1109Noch keine Bewertungen
- Design Considerations of High Voltage AnDokument114 SeitenDesign Considerations of High Voltage AnEL BRIGHLINoch keine Bewertungen
- Unit Hydrograph DerivationDokument7 SeitenUnit Hydrograph DerivationSudharsananPRSNoch keine Bewertungen
- Company Directive: Standard Technique: Sd8A/3 Relating To Revision of Overhead Line RatingsDokument33 SeitenCompany Directive: Standard Technique: Sd8A/3 Relating To Revision of Overhead Line RatingsSathish KumarNoch keine Bewertungen
- Kinematics of Machinery: Motion and AnalysisDokument29 SeitenKinematics of Machinery: Motion and AnalysisShashank SinghNoch keine Bewertungen
- Modelling Urban Areas in Dam-Break Flood-Wave Numerical SimulationsDokument14 SeitenModelling Urban Areas in Dam-Break Flood-Wave Numerical SimulationsDaru Nurisma PramuktiNoch keine Bewertungen
- About The Company: Machined and Forged ComponentsDokument18 SeitenAbout The Company: Machined and Forged ComponentsankitNoch keine Bewertungen
- Physics SL Paper 3 TZ2Dokument20 SeitenPhysics SL Paper 3 TZ2Dongjean SeoNoch keine Bewertungen
- Electric Current and Charge RelationshipDokument9 SeitenElectric Current and Charge RelationshipLokman HakimNoch keine Bewertungen
- Design and Manufacturing of Automatic Gear Shifter For BicycleDokument10 SeitenDesign and Manufacturing of Automatic Gear Shifter For BicycleMannam RujendraNoch keine Bewertungen