Beruflich Dokumente
Kultur Dokumente
Symbols and Units: Script Fonts Are Used For Physical Quantities and in Bold Font
Hochgeladen von
Singh AnujOriginaltitel
Copyright
Verfügbare Formate
Dieses Dokument teilen
Dokument teilen oder einbetten
Stufen Sie dieses Dokument als nützlich ein?
Sind diese Inhalte unangemessen?
Dieses Dokument meldenCopyright:
Verfügbare Formate
Symbols and Units: Script Fonts Are Used For Physical Quantities and in Bold Font
Hochgeladen von
Singh AnujCopyright:
Verfügbare Formate
Symbols
and
Units
Symbols
in
italic
or
script fonts
are
used
for
physical
quantities
and
in
bold
font
for
vectors
or
matrices.
Roman
font
symbols
are
used
for
labels
and
units
e.g.
A
is
the
Helmholtz
free
energy,
but
A
is
a
label
(e.g.
of
the
left
compartment
in
Fig.
1.1);
W
is
the
amount
of
work,
but
W
is
a
unit
of
power
[1
W
=
1
J/s].
For
the
few
symbols
that
have
multiple
meanings
(e.g.,
a),
the
most
common
one
is
listed
first.
SI
units
for
the
corresponding
physical
quantities
are
given
in
brackets
when
appropriate.
Symbols
Latin
alphabet
A
A
a
B
B
b
C
c
D
d
d
E
F
F
f
f
G
g
g
H
h
h
i,
j
K
k
Helmholtz free energy [J (kg m2 )/s2]
area [m2]
activity [dimensionless] ; constant in property relationships [units vary]
second virial coefficient [m3 / mol]
extensive thermodynamic property (e.g. U, V, S, A, H or G) [units vary]
constant in property relationships [units vary]
heat capacity [J/(mol K)]; concentration [mol/m3] ;
third virial coefficient [mol2/ m6]
constant in property relationships [units vary]
diffusion coefficient [m2/ s]
diameter [m]
discriminant of quadratic polynomial
energy [J (kg m2) / s2]
force [N (kg m) / s2]
macroscopic degrees of freedom [integer]
fugacity [Pa 1 kg/(m s2)]
mathematical function
Gibbs free energy [J (kg m2) / s2]
acceleration of gravity [ = 9.81 m/s2 at sea level]
mathematical function
enthalpy [J (kg m2) / s2]
height [m]
mathematical variable or function
species indices [integers]
reaction equilibrium constant [units vary]
spring constant [kg / s2 = N/m]
ii
Symbols and Units
Symbols Latin alphabet (cont.)
k
number of streams [integer]
kB
Boltzmanns constant, R/NA [= 1.380651023 J/K]
kij
interaction parameter in equations of state
l
length or displacement [m]
M
molar mass [kg/mol]
M
matrix of stoichiometric coefficients
m
mass [kg]; chain length [integer]
N
number of moles [mol]
NA
Avogadros number [ = 6.02211023 mol1]
n
number of components [integer]
P
pressure [Pa kg/(m s2) = J/m3; also bar 105 Pa]
P
probability [dimensionless]
p
momentum [kg m/s]
Q
heat [J (kg m2) / s2]
Q
canonical partition function [dimensionless]
q
quality of mixed vapor/liquid stream (fraction vapor) [dimensionless]
q
molecular partition function [dimensionless]
R
Ideal-gas constant [ = 8.3145 J/(mol K)]
R
reactant species (vectors of elemental content)
r
intermolecular distance [nm]; number of independent reactions [integer]
S
entropy [J/K)]
T
temperature [K]
t
time [s]
U
internal energy [J (kg m2) / s2]
U
interaction energy between molecules
u
velocity [m/s]
V
volume [m3, also L 0.001 m3 = 1000 cm3]
W
work [J (kg m2) / s2]
w
mass fraction [dimensionless]
w
mathematical variable or function
X
a chemical species
x
mole fraction [dimensionless] ; horizontal position [m]
x
mathematical variable or function
y
mole fraction [dimensionless]
y
mathematical variable or function
Z
compressibility factor ( PV / RT )
z
vertical position [m]
z
mathematical variable or function
Symbols Greek alphabet
P
expansion ratio [dimensionless]
coefficient of thermal expansion [1/K]
Symbols and Units
iii
Symbols Greek alphabet (cont.)
inverse temperature, 1/kBT [J1]
activity coefficient; ratio of heat capacities CP/CV [dimensionless]
small difference (as prefix) ; solubility parameter [J1/2cm3/2]
difference (as prefix)
energy parameter in intermolecular potential functions [J]
coefficient of performance for cooling cycle [dimensionless, can be > 1]
efficiency of thermal engine [dimensionless, < 1]
thermometric temperature [ C]
T
isothermal compressibility [1/Pa]
multiplication factor [dimensionless]
chemical potential [J/mol]
stoichiometric coefficient of a species [dimensionless]
reaction extent [mol]
a Legendre transform of the fundamental equation [J/mol]
ratio of circumference to diameter of a circle [3.14159]
osmotic pressure [Pa kg/(m s2) = J/m3; also bar 105 Pa]
density [mol/m3 or kg/m3]
distance in intermolecular potentials [m; also 1010 m]
fugacity coefficient [dimensionless]
volume fraction [dimensionless]
number of phases in a system [integer]
Flory interaction parameter [dimensionless]
acentric factor [dimensionless]
number of microstates [dimensionless]
Modifiers
Intensive properties denoted by underscore (e.g. U_ is the energy per mole or kg)
Partial molar quantities denoted by overbar (e.g. Hi is the partial molar enthalpy of
component i)
Rate of change with time or flow rate is denoted by overdot (e.g. !N is the change of
the number of moles per unit time or the flow rate of a stream)
Subscripts and superscripts
constant-pressure property: subscript P
constant-volume property: subscript V
coexistence (phase equilibrium) property: superscript sat
critical-point property: subscript C
excess property: superscript EX
formation property: subscript form
ideal-gas property (omitted if the context is clear): superscript IG
ideal mixture: superscript IM
iv
Symbols and Units
infinite-dilution property: superscript
Subscripts and superscripts (cont.)
liquid phase property: subscript L
mixing property: subscript mix
non-dimensional property: superscript *
normal boiling point property: subscript b
property at the vapor pressure: superscript VP
reaction property: subscript rxn
reduced property: subscript r (e.g. Tr = T/TC)
reference or standard state: superscript 0
reversible process: superscript rev
vapor phase property: subscript V
Braces
{ }
indicates a set of independent mole numbers or mole fractions of compo-
nents in a mixture: { N } means the set of N1, N2, Nn , and { x } means the set
of x1, x2, xn1 .
Abbreviations
FE
QED
rxn
vdW
fundamental
equation
quod
erat
demonstrandum
(Latin
for
which
was
to
be
demonstrated)
reaction
van
der
Waals
Chapter
1.
Introduction
1.1
What
is
Thermodynamics?
The
term
thermodynamics
comes
from
the
Greek
words
o+,
meaning
heat
dynamics
(motion).
Definitions
of
the
term
include:
The
branch
of
physical
science
that
deals
with
the
relations
between
heat
and
other
forms
of
energy
(such
as
mechanical,
electrical
or
chemical
ener-
gy)
and,
by
extension,
the
relationships
and
interconvertibility
of
all
forms
of
energy.
[New
Oxford
American
Dictionary]
The branch of physical science concerned with equilibrium in material
systems and the changes that occur as a result of the interactions with oth-
er systems. [Encyclopdia Britannica Online]
The study of restrictions on the physical properties of matter that follow
from the symmetry properties of the fundamental laws of physics. [Callen]
The
first
two
definitions
contain
terms
that
require
their
own
definitions
(heat
and
equilibrium,
respectively)
while
the
third
is
extremely
general
but
also
a
bit
less
informative.
It
is
clear
from
these
definitions,
however,
that
thermo-
dynamics
is
a
general
tool
applicable
to
many
types
of
physical
systems.
For
exam-
ple,
while
we
will
not
be
dealing
with
systems
of
this
type
here,
thermodynamic
principles
can
be
used
to
analyze
properties
of
black
holes
in
astrophysics.
The
power
of
thermodynamics
is
that
it
generates
useful
relationships
that
are
both
rigorous
and
widely
applicable,
from
a
small
number
of
postulates
supported
by
experimental
observations.
Strong
justification
for
the
postulates
is
provided
by
using
them
to
make
concrete
predictions,
which
are
then
verified
by
comparisons
against
real-world
data.
In
this
book,
the
focus
is
primarily
on
classical
thermodynamics
of
fluids
and
solids
at
conditions
for
which
external
magnetic,
electrical
and
gravitational
fields
have
negligible
influence
on
the
properties
of
the
systems
of
interest.
Some
refer-
ences
will,
however,
be
made
to
the
microscopic
basis
of
thermodynamic
proper-
ties,
even
though
one
can
obtain
all
the
relations
of
classical
thermodynamics
Material
from
Essential
Thermodynamics,
Drios
Press
(2011),
A.Z.
Panagiotopoulos
Hawking,
S.
W.,
Particle
Creation
by
Black
Holes,
Commun.
math.
Phys.,
43:199-220
(1975).
Chapter 1. Introduction
without making any assumptions about the microscopic constitution of matter.
The microscopic point of view is becoming increasingly important in modern en-
gineering, which seeks control of matter at near-atomistic (nm) length scales.
Understanding thermodynamics is of fundamental importance for many
branches of science and engineering. Our point of view in this book is that of chem-
ical engineers, who need to answer questions such as:
How much work is needed to compress a gas?
At a particular temperature and pressure, will a certain liquid vaporize?
What
is
the
solubility
of
a
pollutant
in
water
and
in
the
fat
tissue
of
river
fish?
How far will a chemical reaction proceed in a vessel of known volume
loaded with given amounts of reactants? How can we maximize the yield
of desired products and minimize unwanted side reactions?
How can we modify a protein in order to maximize its thermal stability?
Under what conditions will a certain surfactant form micellar aggregates?
Are two polymers compatible (miscible) with each other?
What
is
the
maximum
amount
of
electricity
that
can
be
obtained
from
a
ge-
othermal
reservoir?
For many of the questions above, other considerations, such as inefficiencies in
mechanical devices, rates of diffusion, and kinetics of chemical reactions often play
a crucial role. However, thermodynamics provides the driving force for any trans-
formation of energy or material condition. It also places hard limits that cannot be
exceeded, irrespective of how clever the design of a specific process might be.
1.2 Thermometric and Ideal-Gas Temperature Scales
The need to quantify the intuitive notion of hot and cold (e.g. boiling water ver-
sus ice) led to the development of simple instruments, such as liquid-in-glass
thermometers that produce an objective measure the length of a liquid column
that correlates with the subjective feeling of warmth. The simple experimental
layout of Fig. 1.1 can be used to confirm that isolated systems tend to approach the
same thermometric temperature when contacted via a thin metal wall. This state-
ment is sometimes referred to as the Zeroth Law of thermodynamics. The two
systems A and B are considered to be in thermal equilibrium after a sufficiently
long time has passed and the thermometric temperatures have become equal, A =
B. It has been empirically determined that two systems in thermal equilibrium
can exist over long periods of time at quite different pressures and compositions, if
the boundary between them is rigid and impermeable to mass.
1.2 Thermometric and Ideal-Gas Temperature Scales
Figure
1.1
Experimental
layout
for
measuring
equality
of
thermometric
tem-
peratures
at
equilibrium.
A thermometric temperature scale can be established by defining a standard
measurement device including its physical dimensions, working fluid (e.g. Hg),
materials of construction (type of glass), as well as at least two reference points.
For example, the two reference points in the commonly used Celsius temperature
scale are (i) the triple-point condition at which pure liquid water coexists with sol-
id ice and water vapor, 1 = 0.01 C and (ii) the boiling point of pure water at at-
mospheric pressure, 2 = 100 C. While it is not obvious a priori that these condi-
tions are reproducible and unique, many careful measurements have established
that they do establish a reliable empirical temperature scale.
A different temperature scale that turns out to play a key role in thermody-
namics is the one provided by the volumetric properties of low-pressure gases.
The key observation here is that the product of pressure and molar volume, P V , for
gases at sufficiently low pressures, is a linear function of thermometric tempera-
Figure 1.2 Volumetric properties of low-pressure gases.
Chapter 1. Introduction
ture, as shown schematically in Fig. 1.2:
PV = a + b
(1.1)
The
linear
relationship
of
Eq.
1.1
is
only
valid
for
a
limited
range
of
thermometric
temperatures
and
breaks
down
at
low
temperatures
(hence
the
dashed
line
in
the
figure).
Setting
the
values
for
constants
a
and
b
effectively
defines
a
thermometric
temperature
scale;
a
particularly
simple
and
appealing
choice
is
b
=
0,
for
which
the
temperature
scale
becomes
the
thermodynamic
temperature
T.
In
this
case,
a
is
identified
as
the
ideal
gas
constant
and
has
the
value
R
=
8.3145
J/(mol
K).
Eq.
1.1
then
becomes:
PV = RT
(1.2)
The relationship between thermodynamic and Celsius temperature scales is:
T [K] = [C] + 273.15
(1.3)
In many problems in this book we will perform calculations to 3 significant figures,
setting T [K] [C] + 273 .
1.3 Systems and Boundaries
A key concept in thermodynamics is the system, which is simply a well-defined re-
gion of space. This region of space does not need to be stationary it can be mov-
ing as a function of time. Once we have decided which region of space we are de-
fining as the system, the environment is defined as everything that is not part of the
system; the surface dividing the system from its environment is called the bounda-
ry. For example, in Fig. 1.1, one could choose as the system of interest compart-
ment A, compartment B, or both compartments taken as a whole. The best system
choice to facilitate thermodynamic analysis of a problem is often not obvious.
However, this choice cannot change the physical properties of the real system be-
ing analyzed, or the results of thermodynamic analysis of the system. For example,
the final temperatures and pressures after a certain physical process has proceed-
ed to equilibrium cannot depend on the (arbitrary) division of the universe into
system and environment.
Systems are characterized by the types of exchanges (flows) that take place
across their boundaries, as follows:
Open / Closed: If system boundaries allow the flow (exchange) of mass be-
tween system and environment, then the system is characterized as open; in
the opposite case we have a closed system. For example, the act of pulling the
ring at the top of a soda can converts it from a closed to an open system.
1.3 Systems and Boundaries
Movable
/
Rigid:
If
system
boundaries
are
deformable,
then
the
shape
of
the
system
can
change
and
its
volume
is
not
constant;
in
the
opposite
case
the
vol-
ume
of
the
system
is
fixed.
Adiabatic
/
diathermal:
Experiments
on
systems
similar
to
the
two-compart-
ment
insulated
container
shown
in
Fig.
1.1
have
demonstrated
that
different
materials
and
wall
thicknesses
result
in
substantially
different
rates
of
tem-
perature
equalization
on
the
two
sides
of
the
partition.
An
idealized
limiting
case
is
that
of
a
partition
that
does
not
allow
any
observable
changes
in
tem-
perature
of
the
compartments,
as
long
as
no
other
energy
input
is
provided
to
them.
In
this
limiting
case,
the
boundary
is
characterized
as
adiabatic
(a
tech-
nical
term
meaning
not
allowing
passage).
The
conceptual
opposite
of
an
ad-
iabatic
boundary
is
denoted
as
diathermal
(e.g.,
a
thin
metal
wall).
A
system
surrounded
by
rigid,
impermeable
and
adiabatic
boundaries
is
isolat-
ed.
This
classification
of
boundaries
seems
somewhat
arbitrary.
How
rigid
does
a
boundary
have
to
be
to
qualify
as
such?
Are
the
thin
walls
of
a
soda
can
sufficient-
ly
rigid?
Even
the
apparently
simple
question
of
whether
a
system
is
open
or
closed
cannot
really
be
answered
unless
some
characteristic
time
of
interest
is
first
defined.
To
illustrate
this
point,
consider
the
following
example.
Example
1.1
A
small,
hermetically
sealed
glass
capsule
contains
He
gas
at
a
pressure
of
P
=
2
bar;
the
environment
is
air
at
atmospheric
pressure.
The
capsule
glass
walls
have
a
thickness
of
0.1
cm.
Is
this
system
closed
or
open?
Clearly,
the
initial
reaction
would
be
to
think
of
a
sealed
glass
capsule
as
a
closed
system.
However,
one
may
recall
that
helium
seems
to
be
able
to
escape
from
apparently
sealed
environments,
such
as
the
interior
of
rubber
party
bal-
loons
(which
deflate
after
a
few
hours).
Analysis
of
this
problem
requires
the
cal-
culation
of
a
characteristic
time
for
diffusion
through
the
l
=
0.1
cm
glass
walls.
The
diffusion
coefficient
of
He
through
glass
is
D
=
8109
cm2/s
at
T
=
300
K.
Dimen-
sional
analysis
(which
simply
means
combining
D
and
l
so
that
the
result
has
units
of
time)
gives
t
=
l
2/ D ,
so
that:
l2
0.1 0.1!cm 2
t= =
= 1.2 10 6 !s! !14!d
9
2
D
8
10
!cm
/s
!
What
can
we
conclude
from
this
calculation?
If
the
time
scale
of
interest
is
much
less
than
14
days,
closed-system
thermodynamic
analysis
is
appropriate.
Closed-
system
analysis
predicts
that
the
final
pressure
inside
the
capsule
remains
at
P
=
Rogers, W. A. et al., "Diffusion Coefficient, Solubility, and Permeability for Helium in
Glass," J. Appl. Phys. 25:868-875 (1954).
Chapter 1. Introduction
2
bar;
this
is
in
agreement
with
observations
at
short
times.
However,
if
the
time
scale
of
observation
is
much
greater
than
14
days,
the
system
can
be
analyzed
as
an
open
system
with
respect
to
the
He
gas.
The
final
pressure
depends
on
whether
sufficient
time
is
allowed
for
diffusion
through
the
glass
walls
of
O2
and
N2
from
the
outside.
The
interior
pressure
is
likely
to
drop
to
a
very
low
value
and
then
eventually
recover
(after
many
years
the
diffusion
of
O2
and
N2
is
much
slower
that
that
of
He)
to
match
the
external
pressure
and
composition.
In
general,
correct
thermodynamic
analysis
will
lead
to
agreement
with
exper-
imental
observations
if
the
assumptions
made
when
performing
the
thermody-
namic
analysis
(closed
/
open
system,
diathermal
/
adiabatic,
etc.)
are
consistent
with
the
real
system
and
time
scale
of
interest.
1.4 Equilibrium States
A basic postulate of thermodynamics is that stable equilibrium states exist for sys-
tems with well-defined external and internal constraints the domain of applica-
bility of classical thermodynamics is precisely these equilibrium states. Such states
are usually obtained after a sufficiently long time has passed so that the observa-
ble properties of systems no longer change with time; it should be emphasized
here that the condition of no change with time is necessary but not sufficient for
characterizing equilibrium states.
The concept of time scales illustrated in the previous section applies here as
well, as it is clear that in order to determine that a property does not change, one
needs to observe it over a certain time scale of interest. One may be tempted to
suggest that the correct thermodynamic equilibrium state of a system is the one
at extremely long times. However, this can lead us down paths that we may not
want to take as engineers try searching the web for Exasecond to obtain an in-
triguing list of transformations that are expected to occur over truly long time
scales. For example, elements lighter than iron are in principle metastable with
respect to nuclear fusion, but these transformations do not take place on any con-
ceivable time scale, except in the interior of stars. Closer to engineering applica-
tions, many chemical reactions proceed extremely slowly at room temperature,
e.g. that of gaseous O2 with H2 in the absence of a catalyst. A mixture of these two
gases can be analyzed as a non-reacting equilibrium thermodynamic system, as
long as no spark or catalyst is present.
Equilibrium
states are char-
acterized by
n+2 independ-
ent variables
For closed systems with no internal boundaries and containing known
amounts of n chemical species (components), equilibrium states are defined as
states that can be fully characterized by exactly 2 thermodynamic variables such
as the temperature, pressure, or overall volume. However, not all choices of varia-
bles are appropriate; for example, temperature and pressure are not independent
variables for a one-component system that contains two coexisting phases. It was
1.4 Equilibrium States
mentioned
in
the
previous
paragraph
that
some
systems
do
not
reach
equilibrium
states
even
after
a
long
time;
a
typical
example
is
common
silicate
glass.
In
a
non-
equilibrium
system,
additional
information,
such
as
the
thermal
history
of
the
ma-
terial,
is
also
necessary
to
characterize
its
state.
Chapter 2. Work, Heat, and Energy
The establishment of heat as a form of energy that can be partially converted into
useful work was a key impetus for the development of thermodynamics as a mod-
ern science in the nineteenth century by Sadi Carnot, James Joule and William
Thompson (Lord Kelvin). The generation of useful work continues to be a funda-
mental underpinning of our technological civilization. For example, we convert
between different forms of energy when powering trains, planes, automobiles, and
boats, when providing a comfortable environment with lighting, cooling, and air
conditioning, or when we perform physical and chemical transformations in
manufacturing. Electricity generation using coal, natural gas, or nuclear fission,
and locomotion using internal combustion engines also depend upon the trans-
formation of heat into other, more useful, forms of energy.
The main objective of the present chapter is to establish the fundamental rela-
tionships between work, heat, and the energy of closed and open systems. Energy
conservation is often called the First Law of thermodynamics. A First Law bal-
ance is usually the starting point for analysis of processes and systems of interest
to this book. Two important quantities, the energy and enthalpy are defined in this
chapter. In order to provide example applications of First Law balances to systems
of common interest in engineering practice, we also discuss energies and enthal-
pies of ideal gases, and how to obtain thermodynamic data for real fluids and solids.
2.1 Mechanical Work
A work interaction between two systems occurs when their boundary moves un-
der the action of a force F. For one-dimensional displacement over a differential
distance dx, the amount of work d/ W is:
/ = Fdx
dW
(2.1)
In
Eq.
2.1,
the
amount
of
work
is
an
inexact
differential
(denoted
by
a
d
with
a
slash
through
it),
meaning
that
the
total
amount
of
work
between
two
specified
posi-
tions
generally
depends
on
the
path,
since
the
force
itself
depends
on
the
path.
The
total
amount
of
work
produced
for
a
finite
displacement
is:
Material
from
Essential
Thermodynamics,
Drios
Press
(2011),
A.Z.
Panagiotopoulos
2.1 Mechanical Work
(2.2)
W = F dx
!
On
the
schematic
diagram
of
Fig.
2.1,
the
force
is
higher
when
the
system
moves
to
the
right
relative
to
when
it
moves
to
the
left.
This
type
of
behavior
is
called
hysteresis
and
occurs
in
sys-
tems
that
have
dissipation
or
friction.
Even
when
well
lubricated,
real
me-
chanical
systems
are
never
completely
free
of
friction;
internal
gradients
of
pressure
or
temperature
also
result
in
Figure
2
.1
F
orce
v
ersus
d
isplacement
f
or
a
hysteretic
behavior.
In 1845, James Joule performed ex-
periments measuring the changes in the
thermometric temperature of an insu-
lated tank of known contents, resulting
solely from mechanical work input. In
particular, a weight was made to de-
scend slowly (Fig. 2.2), producing work
in the form of motion of a stirrer. The
stirring action was shown to increase
the temperature of the working fluid by
an amount directly proportional (for
small temperature changes) to the
amount of work input. This established
that mechanical work could be con-
verted into heat. Through more pre-
cise measurements, the conversion fac-
tor was later established as 4.18 J/g/C
of temperature rise for water at ambi-
ent conditions.
system with hysteresis.
Figure
2.2
Schematic
illustration
of
Joules
experimental
setup
for
conversion
of
mechanical
energy
into
heat.
The
reverse
experiment,
using
a
fluid
to
produce
mechanical
work,
can
also
be
devised.
Such
work
production
is
accompanied
by
a
lowering
of
the
fluids
temper-
ature.
Consider
a
piston
of
mass
m
confining
a
pure
gas
of
temperature
T0
and
pressure
P0
in
an
insulated
cylinder,
with
vacuum
on
the
other
side
of
the
piston
(Fig.
2.3).
Let
us
assume
also
that
mg
<
P0A,
where
A
is
the
cross-sectional
area,
so
that
the
weight
of
the
piston
does
not
balance
the
pressure
from
the
gas.
A
stop
blocks
the
piston
from
moving.
If
the
stop
is
suddenly
removed,
the
gas
will
ex-
pand;
if
there
is
enough
gas,
the
piston
will
reach
the
top
of
the
tank,
as
shown
in
Fig.
2.3,
right
side.
What
is
the
work
interaction
between
the
piston
and
the
gas?
Clearly,
work
is
done
on
the
piston
by
the
gas.
The
net
amount
of
work
needed
to
lift
the
piston
can
be
found
from
Eq.
2.2,
10
Chapter 2. Work, Heat, and Energy
Figure
2.3
Piston
and
cylinder
in
insulated
tank.
W
= F dz = mgh
! on!piston
(2.3)
where
z
is
the
vertical
position.
One
can
also
obtain
the
amount
of
work
performed
on
the
gas
as
it
expands:
W
= PA dz = P dV
! on!gas
(2.4)
The
negative
sign
is
present
here
because
we
use
the
convention
that
work
is
posi-
tive
if
it
is
added
to
a
system
and
negative
if
it
is
produced
by
a
system.
The
pres-
sure
of
the
gas
becomes
lower
as
it
moves
up,
so
that
the
integral
of
Eq.
2.4
re-
quires
some
effort
to
evaluate.
Because
of
imbalance
between
the
force
of
gravity
pulling
the
piston
down
and
the
force
exerted
by
the
gas,
the
piston
will
initially
accelerate
upwards.
However
the
container
wall
eventually
stops
the
piston
and
the
final
equilibrium
state
is
reached
with
the
gas
at
some
Tf
and
Pf .
The
system
can
be
taken
from
the
initial
state
(T0
,
P0)
to
the
same
final
state
(Tf
,
Pf)
in
a
different
way.
Instead
of
letting
the
piston
accelerate
upwards
to
hit
the
top
wall
and
come
to
an
abrupt
stop
there,
a
process
can
be
devised
to
gradually
move
it
up
by
lifting
a
pile
of
sand.
At
the
initial
state
shown
in
Fig.
2.4,
the
weight
of
the
sand
and
piston
just
about
balance
the
pressure
of
the
gas.
By
gradually
re-
Figure 2.4 Piston-cylinder system with gradual lifting.
2.1 Mechanical Work
11
moving
a
few
grains
at
a
time,
the
piston
can
be
lifted
to
the
top
of
the
container.
In
order
to
ensure
that
the
final
state
is
the
same
as
before,
a
large
constant-volume
reservoir
at
Tf
,
the
final
temperature
of
the
experiment
in
Fig.
2.3,
is
placed
in
con-
tact
with
the
gas
when
the
piston
has
reached
the
top.
Since
thermodynamic
sys-
tems
of
one
chemical
component
are
fully
defined
by
n+2
=
3
variables
at
equilib-
rium,
specifying
the
amount
of
gas,
its
final
volume
and
final
temperature
also
sets
the
final
pressure
to
the
same
value
as
previously.
However,
by
the
time
the
piston
has
reached
the
top
of
the
cylinder,
more
work
has
been
performed
by
the
gas
in
this
case
relative
to
that
of
Fig.
2.3.
In
addition
to
lifting
the
piston,
the
gas
has
lift-
ed
some
sand,
as
shown
in
Fig.
2.4.
Where
did
the
extra
work
for
lifting
the
sand
come
from?
It
must
have
come
from
heat
interactions
with
the
reservoir.
This
and
many
other
similar
experiments
establish
that
a
form
of
energy
is
transferred
when
temperature
differentials
exist
between
parts
of
a
composite
system.
When
this
form
of
energy
is
transferred
into
a
substance,
its
temperature
increases.
We
now
know
that
this
energy
is
stored
as
kinetic
and
potential
energy
of
molecules,
but
at
the
time
of
Joules
experiments
the
molecular
hypothesis
for
the
composition
of
matter
was
not
yet
accepted.
However,
for
classical
thermody-
namics,
the
precise
nature
of
this
energy
is
immaterial:
what
is
important
is
that
it
can
be
obtained
from
macroscopic
mechanical
energy
in
a
precisely
quantifiable
fashion.
2.2 First Law for Closed Systems
A key fundamental postulate of thermodynamics is that there is an energy func-
tion E that depends solely on the state of a system. The change in energy, E, be-
tween two equilibrium states of a system such as the one described in the previous
section can be measured directly in experiments involving only mechanical work
interactions. Innumerable attempts by ingenious inventors to construct perpetual
motion machines that could generate energy without any permanent physical or
chemical transformations of their contents have failed. No such devices exist, for
reasons that are strongly entwined with the basic conservation principles of the
physical laws.
Experiments similar to those described in the previous section suggest that
when two systems are in intimate contact, energy that is not associated with mac-
roscopic mechanical forces can flow through their boundary. This hidden energy
needed to balance the books is defined as heat and denoted by the symbol Q. The
amount of heat exchanged between two systems can be measured by first estab-
lishing the energy function of either system using well-insulated (adiabatic) exper-
iments. In its simplest form, written for a closed system that cannot exchange mass
with its environment, the First Law of thermodynamics is written as:
First Law,
closed systems,
integral form
12
Chapter 2. Work, Heat, and Energy
!E = Q +W
work
and
heat
sign
convention
(2.5)
This equation states simply that the change of en-
ergy of a system is due to addition of heat and mac-
roscopic (mechanical) work input. Both work and
heat are considered positive if performed on the
system of interest.
The energy E that appears on the left-hand side
of Eq. 2.5 can have a number of different manifesta-
tions, in particular as potential, kinetic, and internal
energy.
Figure
2.5
Mass
in
a
gravita-
tional
field.
Potential
energy
is
due
to
the
position
of
a
system
in
a
force
field
for
example,
mass
in
a
gravitational
field
or
charge
in
an
electrostatic
potential.
For
a
mass
m
in
a
gravitational
field
(Fig.
2.5),
the
force
is:
(2.6)
!F = mg
The
work
performed
by
the
gravitational
field
when
the
mass
moves
from
height
z1
to
z2
is:
W = 2 mgdz = mg(z1 z2 )
z1
!
(2.7)
If z2 < z1 , work is positive, as it is performed on the system of interest by the ex-
ternal field.
Kinetic energy is the energy associated with macroscopic motion of parts of a
system. The kinetic energy change when a mass m is accelerated from rest (zero
velocity) to a final velocity uf can be found from:
d2x
du
dx
Ekin = Fdx !!!,!!!!where!!F = m 2 = m !!!!!!and!!!!u =
dx = udt
dt
dt
dt
!
u
f
du
1
!!Ekin = m udt = mudu = muf2
dt
2
0
!
(2.8)
The internal energy, denoted by the symbol U, is due to microscopic interac-
tions (intermolecular potential energy) and molecular motions (microscopic kinet-
ic, vibrational, rotational energy). U is an extensive thermodynamic function pro-
portional to the size of a system, U=NU. The molar (specific) energy U for equilib-
rium states of pure components is a function of two independent thermodynamic
variables (such as the temperature and volume). It can be measured through pure-
ly macroscopic observations, by performing an adiabatic change of the condition of
a system and observing the mechanical work input associated with the change. For
13
2.2 First Law for Closed Systems
simplicity, we often omit the internal
adjective and refer to U simply as the
energy of a thermodynamic system.
The energy U is a key thermo-
dynamic quantity and will be central
in later derivations. When written as a
function of the other extensive varia-
bles of a system it becomes a funda-
mental equation for the thermody-
namic state of a system, as discussed
in detail in Chapter 5 of this book. We
Figure 2.6 Extension of a linear spring.
can write a differential form of the
First Law by considering changes to
the internal energy of a system resulting from the addition of small amounts of
work and heat:
/ + dW
/
!dU = dQ
(2.9)
In
writing
Eq.
2.9,
we
have
assumed
that
there
are
no
changes
in
kinetic
or
poten-
tial
energy
of
the
system.
Even
though
the
energy
is
a
function
of
thermodynamic
state
(hence
it
has
a
proper
differential
dU),
both
heat
and
work
terms
depend
on
the
path
taken
and
are
thus
represented
by
inexact
differentials.
In
some
cases,
the
distinction
between
internal
and
potential
energy
can
be
somewhat
arbitrary.
For
example,
a
simple
linear
spring
(Fig.
2.6),
exerts
a
force
on
its
environment
that,
for
small
extensions,
follows
the
equation
F
=
k(x
x0),
where
x
is
the
instantaneous
position
of
the
end
of
the
spring
and
x0
is
the
length
at
rest.
The
energy
change,
E,
of
the
spring
is
equal
to
the
work
input
into
the
sys-
tem
when
the
spring
end
moves
from
position
x0
to
x1:
x
x
k
(2.10)
E = W = 1 F dx = 1 k(x x0 )dx E = (x1 x0 )2
x0
x0
2
!
!
This
change
in
energy
can
be
considered
to
be
the
change
in
potential
or
internal
energy
of
the
spring,
depending
on
whether
we
adopt
a
macroscopic
(mechanical)
or
molecular
point
of
view.
Example 2.1 A stretched rubber band
A rubber band follows the force-extension relationship of a linear spring. Consider
a stretched rubber band of length x = 0.20 m, with equilibrium (unstretched) length
x0 = 0.10 m and k = 500 N/m (coordinates are as in Fig. 2.6). The rubber band is at
room temperature. Now we allow the band to perform work by returning it to the
unstretched state, x = x0. The release takes place quickly enough so that no appre-
ciable heat transfer to the environment occurs during the release. The heat capaci-
First
Law,
closed
systems,
differential
form
14
Chapter 2. Work, Heat, and Energy
ty, C = (U / T)l of the rubber band is 5 J/K, approximately independent of length.
Estimate the temperature change of the rubber band upon release.
The amount of work performed on the rubber band is given by Eq. 2.10,
W =
x0
F dx =
x0
k
k(x x0 )dx = (x x0 )2
2
500
N
W =
(0.2 0.1)2 ! m2 = 2.5!J
2
m
!
The
work
is
negative
because
the
rubber
band
has
produced
work
on
the
envi-
ronment
when
the
end-point
was
moved.
From
a
First
Law
balance,
U = Q +W U = CT = W T =
W 2.5!J
=
C 5!J/K
T = 0.5!K
!
!
The
quickest
way
to
detect
a
temperature
change
for
a
rubber
band
being
released
from
a
stretched
state
is
to
use
your
lips
feel
free
to
try
this
experiment
at
home,
using
appropriate
eye
protection.
The
potential
energy
of
the
rubber
band
was
considered
part
of
its
internal
ener-
gy
U
in
this
example.
This
is
because
the
elastic
response
is
due
to
molecular-level
changes
within
the
rubber
band.
Example
2.2
Work
and
Heat
of
Vaporization
Calculate
the
work
and
heat
requirements
for
the
process
of
vaporization
of
1
mol
of
liquid
water
from
a
closed
container
at
two
temperatures,
T
=
298
K
and
373
K.
Vaporization
for
pure
components
takes
place
at
constant
pressure,
called
the
va-
por
pressure
in
vaporization,
heat
input
is
not
accompanied
by
a
temperature
increase,
only
a
change
of
physical
state.
Properties
of
water
and
other
substances
can
be
obtained
from
the
NIST
Chemistry
WebBook,
as
explained
in
Appendix
I.
The
environment
is
at
atmospheric
pressure,
Patm
=
101
kPa.
Transformation
of
liquid
into
vapor
is
accompanied
by
a
large
volume
change,
so
the
closed
container
needs
to
have
flexible
walls
or
be
equipped
with
a
movable
piston
to
allow
for
expansion,
as
shown
in
Fig.
2.7.
The
vapor
pressure,
liquid
and
vapor
energies,
and
volumes
at
298
K
are
obtained
from
the
NIST
WebBook
as:
P VP = 3.1417!kPa!
U L = 1.8772!kJ/mol;!!!U V = 43.397!kJ/mol
V = 0.0181!l/mol;!!V V = 787.36!l/mol
! L
An
issue
to
be
considered
in
this
problem
is
the
work
to
push
back
the
atmos-
phere.
Clearly,
the
piston
experiences
a
different
pressure
on
the
left,
!P VP ,
than
on
15
2.2 First Law for Closed Systems
the right, Patm ; a net force to the right is re-
!
quired to keep the piston in place; it is equal
to:
F = (Patm P VP )A ,
!
where
A
is
the
area
of
the
piston.
The
force
is
constant
as
the
liquid
vaporizes
because
the
pressures
on
the
two
sides
of
the
piston
do
not
change.
The
net
work
required
to
move
the
piston
from
the
initial
to
the
final
volume
is
Wtotal = Fx = (Patm P VP )A x =
(Patm P VP )V =
= (101 3.14)103 Pa
!!!!(787.36 0.0181)10 m /mol!
W
= !77.05!kJ/mol
! total
Figure
2.7
Vaporization
in
a
closed
container
(volumes
a re
not
drawn
to
scale).
However,
this
work
is
not
equal
to
the
work
performed
on
the
system
most
of
it
is
performed
on
the
atmosphere.
The
work
performed
on
the
system
is
Wsys = P VPA x = P VP V = 2.47!kJ/mol
!
W is negative because expansion of the system means it performs work on its
! sys
environment.
First Law balance for the closed system now gives:
U = Q +Wsys Q = U Wsys = U V U L Wsys = (41.52+ 2.47)!kJ/mol = 43.99!kJ/mol
!
At
the
normal
boiling
point
(T
=
373
K),
the
total
work
required
Wtotal
=
0,
as
the
pressures
on
the
two
sides
of
the
piston
are
equal.
The
heat
required
is:
Q = U +Wsys = U + P VP V =
= (45.14 7.54)!+101(30.27 0.02)103 kJ/mol =
!= (37.60+ 3.05)!kJ/mol = 40.65!kJ/mol
The large change in internal energy on vaporization of a liquid is due to the need to
disrupt the attractive interactions acting between its molecules, which include hy-
16
Chapter 2. Work, Heat, and Energy
drogen
bonds
in
the
case
of
water.
As
seen
in
this
example,
the
internal
energy
change
is
an
order
of
magnitude
greater
than
the
P VPV
term.
2.3 First Law for Open Systems
Many systems of interest have bound-
aries that are open to the flow of mass.
For example, modern chemical plants
typically operate using continuous-flow
process units, each with one or more
input and output streams. For open sys-
tems, we clearly need to consider the
energy carried by the streams entering
and leaving the control volume when
performing an energy balance. Consider
Figure 2.8 Open system.
an open system, into and out of which
with i = 1 ,2,,k, as shown schematically in Fig.
streams flow at (mass) rates m
! i
2.8. Only three streams are shown in the figure for simplicity. The system is de-
fined as the contents of the box outlined by the heavy black line and dashed lines
at the entrance points it does not include the material in the pipes leading into
and out of the container. The instantaneous mass of the system itself (which could
be changing with time) is m. For simplicity, we will assume that the whole system
has a single velocity u and vertical position z. The analysis can be easily general-
ized for systems with parts that have different velocities and vertical positions.
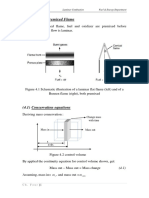
Conservation of energy can be generally written as
u2
!!!!!!!Rate!of!energy
d
U + m + gz =
dt
2
net!input!to!the!system
(2.11)
ui2
Emass!flow = m i U i + + gzi
2
i=1
!
(2.12)
!
The
energy
net
input
to
the
system
contains
heat
and
work
terms,
as
well
as
flow
terms.
Work
and
heat
flows
are
represented
by
arrows
in
Fig.
2.8.
The
rate
of
work
input
into
the
system
is
denoted
by
W
;
this
work
input
could
be
in
the
form
of
elec-
trical
energy,
or
mechanical
work
through
the
action
of
pumps,
compressors,
or
pistons.
The
rate
of
heat
input
into
the
system
is
denoted
by
Q
.
The
rate
of
energy
input
due
to
the
streams
flowing
in
and
out
of
the
system,
assuming
that
the
quan-
tities
m
i
are
algebraic
mass
flow
rates
(positive
for
input
streams
and
negative
for
output),
is:
Ui
is
the
specific
energy
(on
a
mass
basis)
of
stream
i.
However,
there
is
one
addi-
tional
energy
input
that
is
not
as
obvious.
It
has
to
do
with
the
work
required
to
17
2.3 First Law for Open Systems
push the input streams into the system
and the work produced by the output
streams as they come out. The easiest
way to understand this PV term is to
consider a simple system with one in-
put and one output stream (Fig. 2.9)
and a small interval of time during
which mass min enters the system and
mass mout leaves the system. Their
pressures are, respectively, Pin and Pout.
Now consider the closed system that Figure 2.9 PV work in an open system with
a single input and output stream.
consists of the original open system,
augmented by Vin = Vinmin and exclu-
ding Vout = Voutmout. The boundaries of this new system are shown by dotted lines
in Fig. 2.9. The volumes occupied by the masses min and mout may be very differ-
ent, if there are changes in the physical state of the material going through the sys-
tem (e.g. liquid going to vapor, gas being compressed or expanded). Now it should
be clear that the work term associated with the process of pushing min into the
system and getting mout out of the system is:
E
= Pin Vin PoutVout = Pin V!in min Pout V!out mout
! PV !work
in Pout V!out m
out
E
= PinV!in m
! PV !work
(2.13)
Combining Eqs. 2.11 through 2.13,
u2
U
+
m
+ gz = Q + W + Emass!flow + E PV !work
dt
2
u2
i U i + Pi V i + i + gzi
= Q + W + m
2
i=1
(2.14)
!
The
combination
U
+
PV
is
defined
as
a
new
thermodynamic
function
called
the
enthalpy
H.
In
Chapter
5,
we
will
show
that
the
enthalpy
is
also
a
fundamental
equation
with
the
same
information
content
as
the
energy
U.
At
the
moment,
however,
we
consider
this
as
just
a
convenient
way
to
write
the
energy
balance
equation
for
an
open
system.
The
final
result
is:
u2
ui2
d
i H i + + gzi
U + m + gz = Q + W + m
dt
2
2
i=1
(2.15)
18
Chapter 2. Work, Heat, and Energy
Often,
the
kinetic
and
potential
energy
terms
are
small;
if
these
terms
can
be
ignored,
the
open-system
energy
balance
becomes
(now
written
on
a
molar
basis):
First
Law,
dU
open
systems,
= Q + W + H in N in H out N out
(2.16)
dt
diff.
form,
entering
leaving
streams
streams
!
no
kinetic
or
potential
en-
In
Eq.
2.16,
we
have
split
the
sum
over
all
input
and
output
streams
into
two
ergy
changes
terms,
the
first
a
summation
over
entering
streams,
N in ,
and
the
second
over
leav-
!
ing
streams,
N out ,
with
all
flows
taken
as
positive.
Another
useful
way
to
write
Eq.
!
2.16
is
by
multiplying
all
terms
by
dt
to
obtain:
/ + dW
/ +
dU = dQ
entering!
streams
H in dN in
leaving!
streams
H out dN out
(2.17)
!
For
steady-state
operation,
the
left-hand
side
of
Eqs.
2.16
or
2.17
(the
change
of
energy
for
the
contents
of
the
open
system
of
interest)
is
zero.
A
further
simpli-
fication
can
be
obtained
for
the
very
special
case
of
an
open
system
at
steady
state,
which
has
only
a
single
stream
of
flow
rate
N
entering
and
leaving.
For
this
highly
restrictive
case,
the
First
Law
balance
takes
a
particularly
simple
form,
which
may
be
familiar
to
you
from
Materials
and
Energy
Balances
or
other
introductory
courses:
Q + W
N H out H in = NH
= Q + W H =
(2.18)
N
!
If
the
properties
of
entering
and
leaving
streams
do
not
vary
over
the
time
pe-
riod
of
interest,
Eq.
2.16
can
be
integrated
to
give
the
analog
of
Eq.
2.5:
First
Law,
open
systems,
integral
form,
no
kinetic
or
potential
en-
ergy
changes
U = Q +W +
entering
streams
Nin H in
leaving
streams
Nout H out
(2.19)
As
previously,
for
a
steady-state
process,
the
change
in
the
contents
of
an
open
sys-
tem
is
zero,
so
the
left-hand
side
of
Eq.
2.19
vanishes.
It
is
important
here
to
discuss
the
various
forms
of
the
open-system
First
Law
balance
that
have
just
been
presented.
For
example,
Eqs.
2.18
and
2.19
may
at
first
appear
to
be
quite
confusing,
given
that
2.18
has
a
change
of
enthalpy
on
the
left-
hand
side,
while
2.19
has
a
change
of
energy.
However,
the
change
of
enthalpy
in
Eq.
2.18
is
of
the
material
flowing
through
the
open
system,
while
the
change
of
energy
of
Eq.
2.19
is
of
the
open
system
itself.
Eq.
2.18
relies
on
several
assump-
tions
(steady
state,
single
input-output),
while
2.19
is
more
general,
assuming
only
constant
properties
of
the
entering
and
leaving
streams
over
the
period
of
interest.
Of
course,
the
most
general
form
of
the
First
Law
balance
for
open
systems
is
Eq.
19
2.3 First Law for Open Systems
2.15,
which
does
not
require
any
assumptions
this
also
makes
it
the
most
diffi-
cult
expression
to
use
in
practice.
Example
2.3
Vaporization
in
an
open
system
Repeat
the
calculations
of
Example
2.2,
now
considering
a
steady-state
flow
pro-
cess
producing
1
mol/s
of
steam
from
liquid
water
at
its
vapor
pressure.
Let
us
consider
a
flow
process
(Fig.
2.10)
with
input
and
output
streams
at
the
conditions
of
Example
2.2.
Since
input
and
output
streams
are
at
the
same
pressure,
no
pumping
is
required
to
operate
this
process
and
the
work
input
is
zero.
A
steady-state
differ-
ential
First
Law
balance
gives:
dU
V
= Q + W + NH L NH
dt
Q = N H V H L
!
Figure
2.10
Vaporization
in
an
open
system.
We obtain values for the enthalpy
at saturation of water at 298 and 373 K and sub-
stitute in the expression above:
mol
kJ
kJ
45.87 1.88
= 43.99 !!at!298!K!!
s
mol
s
mol
kJ
kJ
Q =1
48.20 7.54
= 40.66 !!at!373!K
s
mol
s
!
Q =1
The
amounts
of
heat
required
for
the
open-system
process
are
equal
(within
nu-
merical
precision)
to
the
corresponding
quantities
for
the
closed-system
process
of
Example
2.2;
there
is
no
real
difference
between
the
two
processes
other
than
accounting
for
the
P VPV
term.
When
considering
a
closed-system
process,
this
term
appears
as
work
explicitly
produced
by
the
expanding
system.
In
an
open
system,
this
work
is
contained
in
the
enthalpy
of
the
flowing
exit
stream.
Example
2.4
Thermodynamic
Analysis
of
the
Human
Heart
A
simplified
schematic
diagram
of
the
human
heart
is
shown
in
Fig.
2.11.
Blood
from
the
body
enters
the
heart
in
point
A,
at
pressure
PA
=
0.66
kPa
above
atmos-
pheric.
The
pressure
difference
from
the
ambient
pressure
is
defined
as
gauge
Inspired
by
problem
13.10
in
Levenspiel,
O.,
Understanding
Engineering
Thermo,
Prentice-
Hall (1996).
20
Chapter 2. Work, Heat, and Energy
pressure.
Blood
exits
the
right
ventricle
in
point
B
on
its
way
to
the
lungs,
at
gauge
pressure
PB
=
2.9
kPa.
Oxygenated
blood
returns
from
the
lungs
in
point
C
at
gauge
pressure
PC
=
1.0
kPa
and
is
in
turn
returned
to
the
circulatory
system
through
ar-
tery
D
at
gauge
pressure
PD
=
13
kPa.
The
normal
blood
flow
rate
(at
rest)
is
3
!V = 90!cm /s .
The
diameters
of
the
veins
and
arteries
are
dA
=
3.0
cm,
dB
=
2.5
cm,
dC
=
2.8
cm
and
dD
=
2.0
cm.
Calculate
the
minimum
power
required
for
operation
of
the
heart,
assuming
no
heat
effects
(!Q = 0 )
and
a
constant
density
of
blood,
=
1
g/cm3.
Since
it
is
not
clear
that
we
can
neglect
kinetic
energy
terms,
we
start
with
a
full
First
Law
bal-
ance
(Eq.
2.15).
At
steady
state,
the
time
differ-
ential
on
the
left-hand
side
of
Eq.
2.15
is
zero.
We
also
do
not
have
significant
potential
energy
changes,
as
the
arteries
and
veins
all
enter
the
heart
at
roughly
the
same
height,
so
gravitation-
al
energy
changes
can
be
neglected.
4
u2
0 = Q + W + m i U i + Pi V i + i + gzi
2
i=1
(i)
Figure
2.11
Schematic
diagram
of
circulation
in
the
human
heart.
We are not given any information on the specific
energies Ui, but since blood going through the
heart does not change when flowing in the atria and out the ventricles, we must
have
4
m
iUi = 0
!i=1
We
can
obtain
the
velocities
from
the
relationship:
2
D
V = uA = u
4
!
u=
4V
2
; uA =
490!cm3 /s!
2
3.14! !3.0 cm
= 12.7
cm
m
= 0.127
s
s
m
m
m
Similarly,
we
get
uB = 0.183! , uC = 0.146!
and
uD = 0.286! .
s !
s
s
!
!
= V = V /V ,
we
obtain:
Now
substituting
in
Eq.
(i),
and
using
!m
4
u2
u2 u2 + u2 u2
i Pi V i + i = V PA PB + PC PD m
A B C D
W = m
2
2
i=1
21
2.3 First Law for Open Systems
cm3
m3
W = 90
106
0.66 2.9+1.013 103 Pa
s
cm3
2
2
2
2
kg 0.127 0.183 + 0.146 0.286 m2
J
0.09
W = (+1.282+ 0.004) = 1.286!W
2
s
2
s
s
!
Most
of
the
work
is
needed
for
increasing
the
pressure
(PV
term);
the
change
in
kinetic
energy
of
the
blood
is
negligible.
The
overall
efficiency
of
mammalian
hearts
is
about
15%,
so
the
actual
power
requirement
is
around
9
W
this
is
the
chemical
energy
that
needs
to
be
provided
in
the
form
of
ATP
to
the
muscle
tissue
driving
the
heart.
The
energy
requirement
per
day
is
9
W86400
s/d
800
kJ/d
190
kcal/d.
2.4 Ideal Gases
When performing quantitative energy balance calculations, it is necessary to have
information on how the pressure and temperature of the system of interest influ-
ence properties such as the density = N/V and internal energy U. Detailed discus-
sion of such equations of state can be found in Chapter 7. Here, we derive some
basic relationships for a special class of substances, ideal gases. In ideal gases, in-
teractions between molecules can be neglected. Real gases at low pressures ap-
proach this condition as the average distance between molecules becomes much
greater than the range of intermolecular forces. Using statistical mechanical meth-
ods introduced in Chapter 5, it can be shown that the pressure P of a system of N
non-interacting particles in a volume V is given by the relationship (see Example
5.8, p. 102, for a derivation):
(2.20)
!PV = NRT
R is called the ideal-gas constant and has the value R = 8.3145 J/(mol K). Since ide-
al gases consist of isolated molecules that do not feel the presence of each other,
their internal energy is simply the sum of energies of the individual molecules and
does not depend on density or pressure:
IG
IG
!U = NU (T )
(2.21)
IG
Here,
we
have
written
!U (T )
to
emphasize
that
the
molar
energy
in
the
ideal
gas
state
is
a
function
of
temperature.
The
internal
energy
of
ideal
gases
contains
contributions
from
kinetic
energy
of
translation
of
the
molecules
in
space,
as
well
as
rotational
and
vibrational
mo-
tions.
It
is
a
function
of
temperature
because
molecules
travel
faster
and
rotational
and
vibrational
motions
store
more
energy
at
higher
temperatures.
22
Chapter 2. Work, Heat, and Energy
Now
let
us
consider
heating
an
ideal
gas
in
a
container
with
rigid
walls
(so
that
no
work
is
performed,
d/W
=
0 ).
Assuming
that
we
can
neglect
the
walls
heat
capac-
ity,
we
can
write
from
a
differential
First
Law
balance
on
this
closed
system:
/ = NCVIGdT
dU IG = dQ
!
We
have
defined
a
new
quantity,
the
heat
capacity
at
constant
volume:
(2.22)
IG
dU
(2.23)
C
dT
!
The
heat
capacity
of
ideal
gases
can
be
measured
experimentally
quite
accu-
rately
for
relatively
simple
systems
it
can
also
be
predicted
theoretically
to
a
high
level
of
accuracy.
For
example,
for
simple
monoatomic
gases
(e.g.
He
or
Ar)
the
heat
capacity
at
constant
volume
is
IG
V
C IG = 3R /2!!(monoatomic!gases)
! V
(2.24)
In general, each fully excited degree of freedom for a molecule contributes
R/2 to the constant-volume heat capacity for monoatomic gases the degrees of
freedom are translational motion in each of the three directions of space. Rotations
and vibrations provide additional degrees of freedom for more complex, polyatom-
ic molecules. While for monoatomic gases the ideal-gas heat capacity is independ-
ent of temperature, for more complex molecules the various degrees of freedom
become excited gradually as temperature rises and the ideal-gas heat capacity is a
function of temperature (see 7.2, pp. 139-141).
As density is increased, real systems deviate from the limiting ideal gas behav-
ior as the molecules feel the presence of each other and their intermolecular in-
teractions start contributing to the total energy. The internal energy per mole, U,
becomes a function of the density in addition to the temperature. In this general
case, we define the heat capacity as the partial derivative of the molar energy with
respect to temperature at constant volume:
U
CV
(2.25)
T V
!
Constant
volume
properties
are
sometimes
called
isochoric.
Heat
capacities
of
real
systems
can
be
measured
directly
and
can
also
be
determined
indirectly
from
the
ideal-gas
values
and
the
volumetric
properties.
Now
let
us
consider
the
case
of
heating
an
ideal
gas,
but
at
constant
pressure
rather
than
constant
volume.
A
differential
First
Law
balance
on
a
container
fitted
with
a
piston
to
maintain
constant
pressure
gives:
/ + dW
/ = dQ
/ PdV
!dU = dQ
23
2.4 Ideal Gases
We
have
used
Eq.
2.2
to
obtain
d/W,
with
the
force
F
=
PA,
so
that
Fdx
=
PdV.
Since
pressure
is
constant,
we
can
rearrange
this
expression
to
obtain:
!U = Q PV Q = (U + PV ) = H [constant-P]
(2.26)
In strict analogy to Eq. 2.23, we can define the constant-pressure (isobaric)
heat capacity for an ideal gas as:
IG
C PIG
dH
d IG
d IG
=
U + PV =
U + RT = CVIG + R
dT
dT
dT
(2.27)
!
As
seen
in
Eq.
2.27,
for
ideal
gases,
the
relationship
between
the
heat
capacities
at
constant
pressure
and
constant
volume
is
particularly
simple.
The
heat
capacity
at
constant
pressure
for
ideal
gases
is
a
function
of
temperature
only
(not
density
or
pressure),
just
as
was
the
case
for
the
heat
capacity
at
constant
volume,
C IG
V .
In
the
general
case,
for
systems
that
cannot
be
described
as
ideal
gases,
the
heat
ca-
pacity
at
constant
pressure
is
again
defined
as
the
temperature
derivative
of
en-
thalpy:
H
1 H
(2.28)
CP =
=
N T N ,P T P
!
In
general,
the
heat
capacity
at
constant
pressure
is
greater
than
the
heat
ca-
pacity
at
constant
volume,
but
the
relationship
between
the
two
is
a
non-trivial
function
of
the
volumetric
properties
of
the
system.
Adiabatic
expansion
of
ideal
gases
follows
simple
expressions
that
are
useful
for
analysis
of
many
physical
systems.
The
derivations
below
are
valid
only
for
ideal
gases.
The
differential
change
of
energy,
assuming
uniform
pressure
P
during
the
expansion
and
no
change
in
kinetic
or
potential
energy
is:
/ + dW
/ = PdV
dU = dQ
!
The
change
in
energy
can
also
be
obtained
from
Eq.
2.22
(with
N
constant):
RT
dU = PdV NCVIGdT = NPdV = NPd
!
RT
CVIGdT = PdV = RdT +
dP
P
!
(2.29)
The
relationships
of
Eq.
2.29
can
be
integrated
in
three
different
ways,
depending
on
the
selection
of
independent
variables.
In
particular,
eliminating
pressure
from
the
first
equation
and
integrating
from
initial
conditions
(labeled
0)
to
a
final
state
(unlabeled)
we
obtain:
24
Chapter 2. Work, Heat, and Energy
RT
dT
dV
CVIGdT = PdV CVIGdT =
dV CVIG
= R
V
T
V
!
V
T V CVIG V
=
= =
(2.30)
T0 V 0
V0
V0
!
In
writing
the
last
equality
in
Eq.
2.30,
we
have
taken
into
account
the
fact
that
the
number
of
moles
N
is
constant
in
this
process,
so
that
ratios
of
specific
and
total
volumes
are
the
same.
We
have
also
defined
the
ratio
of
the
heat
capacities
as
= C P /CV .
Eliminating
volume
from
Eq.
2.29
and
integrating
we
obtain:
!
RT
dT
dP
dT
dP
CVIGdT = RdT +
dP (CVIG + R)
=R
C PIG
=R
P
T
P
T
P
!
R
IG
T P CP P
= =
T0 P0
P0
!
(2.31)
Finally, eliminating temperature from Eq. 2.29 and integrating we obtain:
PdV = C dT =
IG
V
CVIG
VdP =
! R
CVIG
R
IG
CV + R
R
P V
=
P0 V 0
d(PV ) =
CVIG
R
(PdV +VdP)
dP
dV
PdV C
= C PIG
P
V
IG
V
V
=
V0
PV = const.
(2.32)
Once
again,
N
is
constant
in
this
process,
so
that
ratios
of
specific
and
total
volumes
are
the
same.
The
amount
of
work
performed
on
an
ideal
gas
during
adiabatic
compression
can
be
obtained
from
an
integral
First
Law
balance,
if
the
heat
capaci-
ty
is
independent
of
temperature:
U = Q +W W = NCVIG T
!
(2.33)
Example 2.5 Filling of a gas cylinder
An insulated rigid vessel of volume V initially contains Ni moles of an ideal gas at
temperature Ti. A pipe is now connected to the vessel and the same gas at temper-
ature Tin is pumped in, until the number of moles in the vessel becomes Nf. Obtain
an explicit expression for the final temperature in the vessel, Tf, in the form Tf = f (Ti
, Tin , Nf / Ni , CP). CP is the molar heat capacity of the gas at constant pressure, as-
25
2.4 Ideal Gases
sumed to be independent of temperature. There is no heat transfer between the
gas in the tank and the vessel walls during the transfer.
We start with a First Law balance on the contents
of the vessel at a time when there are N moles of
gas in it at temperature T, taking into account that
both heat and work terms are zero:
dU = H in dNin d(UN) = H in dNin
!
Figure
2.12.
Vessel
being
filled
UdN + NdU = H in dNin = Hin dN
!
(i)
Note
that
dU
and
N
refer
to
the
vessel
and
are
changing
as
it
is
being
filled,
while
Hin
and
dNin refer
to
the
incoming
stream,
which
has
constant
properties.
For
this
particular
example,
dN
= dNin
because
there
is
only
one
input
stream
and
any
change
in
the
contents
of
the
vessel
comes
from
that
stream;
this
is
not
generally
the
case.
Since
the
heat
capacity
CP
is
independent
of
temperature
(and
thus
also
is
CV
=
CP
R),
we
can
express
the
enthalpy
and
energy
at
any
temperature
T
in
terms
of
reference
values
H0
and
U0
at
temperature
T0:
T
U(T )U 0 (T0 ) = CV dT U(T ) = U 0 (T0 )+ CV (T T0 )
T0
H(T ) H 0 (T0 ) = C P dT H(T ) = H 0 (T0 )+ C P (T T0 )!,!with!!H 0 (T0 ) = U 0 (T0 )+ RT0
T0
!
A
convenient
choice
for
First
Law
balances
involving
ideal
gases
is
to
set
T0
=
0 and
U0
=
0,
which
results
in
U(T)
=
CVT
and
H(T)
=
CPT.
Even
though
such
a
choice
is
nev-
er
made
in
thermodynamic
tables
of
real
fluids
because
the
absolute
zero
in
tem-
perature
cannot
be
reached,
any
observable
quantities
(such
as
the
temperature
of
the
vessel
in
this
example)
are
independent
of
this
choice
of
reference
states.
Substituting
U
and
H
into
expression
(i)
above
and
using
=
CP/CV
,
we
obtain:
CV TdN + NCV dT = C PTin dN NCV dT = C PTin CV T dN
!
Nf
T
N
T T
dN
dT
= f
ln f = ln in f
Ti T T
N
Ni
Tin Ti
in
N
Tf = Tin i (Tin Ti )
Nf
!
2.5 Properties of Real Fluids
In general, gases deviate from ideal-gas behavior at most conditions of practical
interest. Liquids and solids are, of course, far from being ideal gases at all condi-
26
Chapter 2. Work, Heat, and Energy
tions; their thermodynamic properties cannot be obtained at all from application
of the relationships of the previous section. Directly measurable thermodynamic
properties of real systems, such as the pressure at a given density and tempera-
ture, are instead obtained from careful experiments. The measurements are fre-
quently smoothed and fitted to mathematical expressions for use in calculations.
Derived properties such as the energy U or enthalpy H cannot be measured
directly. They are constructed from the mathematical expressions of the measured
properties using the methods described in Chapter 7. Such properties require the
specification of a reference state at which they are arbitrarily set to zero, because
only differences in derived properties between two thermodynamic states can be
obtained the absolute values of derived properties at a single state cannot be
determined and are of no value for making predictions about measurable quanti-
ties.
High quality, evaluated thermodynamic data for many pure components are
now available from online data sources, as described in Appendix I.
Example 2.6 Expansion of gas in a closed system
Consider the piston-cylinder system of Fig. 2.13. The gas is CO2 and the cylinders
cross-sectional area is A = 0.1 m2. The initial temperature is T0 = 300 K and the ini-
tial pressure is P0 = 2 MPa. The mass of the piston is m = 40 kg and the initial height
of the bottom compartment is h0 = 0.20 m. The height of the gas compartment dou-
bles when the piston reaches the top of the tank, h = h0. Calculate the final temper-
ature Tf and pressure Pf , assuming that there is no heat exchange between the in-
terior of the piston and the outside world walls. This assumption is reasonable for
rapid expansion, during which there is not enough time for appreciable heat trans-
fer to occur between the interior and exterior of the cylinder.
We first obtain the molar
density and energy of CO2
at T0 = 300 K, P0 = 2 MPa
from the NIST WebBook:
0 = 895.71 mol/m3
and U0 = 19.260 kJ/mol.
The number of moles of
gas in the cylinder is
Figure 2.13. Piston-cylinder system
N = h0A 0 = 0.20 m 0.1 m2 895.71 mol/m3 = 17.914 mol
A First Law balance between initial and final states gives:
m
U = Q +W = mgh = 40!kg! !9.81! ! !0.20!m!= 78.48!J
s2
!
27
2.5 Properties of Real Fluids
so that
kJ
! !0.078!kJ!=!344.95!kJ
Uf = U0 + U = ! 17.914!mol! !19.260!
mol
U
344.95!kJ
kJ
= 19.256
The
final
molar
energy
is:
U f = f =
N 17.914!mol
mol
!
The
final
density
is
half
the
initial,
f
=
0.5895.71
mol/m3
=
447.86
mol/m3.
From
the
known
internal
energy
and
density,
an
isochoric
query
of
the
NIST
tables,
with
some
trial-and-error
to
find
an
appropriate
temperature
range,
gives:
Tf = 289.60 K and Pf = 1.0159 MPa
If we had neglected the work term in the First Law balance, we would have ob-
tained Uf = U0 = 19.260 kJ/mol, from which Tf = 289.73 K and Pf = 1.0164 MPa. The
effect of raising the weight of the piston on the final temperature and pressure is
relatively small.
How different would our answer be if we were to assume ideal-gas behavior? The
constant-volume heat capacity of CO2 is CVIG = 28.91 J/(mol k). The final tempera-
!
ture and pressure can be calculated from:
U = NCVIG T Tf = T0
Tf = 300
mgh
NCVIG
= T0
RT0mgh0
PoAh0CVIG
8.314!J/(mol!K)! !300!K! 40!kg! !9.81!m/s2
2106 !J/m3 0.1!m2 28.91!J/(mol!K)
= T0
RT0mg
PoACVIG
= 299.83!K!!
V T
1 299.83
Pf = P0 0 f = 2!MPa
= 0.9994!MPa
Vf T0
2 300
!
The
error
under
the
assumption
of
ideal
gases
is
about
10
K
relative
to
the
more
accurate
calculation
given
earlier.
The
final
pressure,
on
the
other
hand,
is
within
2%
of
the
value
obtained
from
accurate
volumetric
data.
2.6 Problem-Solving Strategies and Additional Examples
Thermodynamics problems can seem quite daunting at first. A common complaint
is that it is not clear how the general principles apply to the specific problem at
hand. In general, one should follow a systematic approach with these steps:
28
Chapter 2. Work, Heat, and Energy
1. Draw a schematic diagram of the process and label key quantities.
2. Define the thermodynamic system(s) of interest.
3. State all assumptions.
4. Write down fundamental thermodynamic relationships (e.g. First Law bal-
ance) for the system.
5. Write down constitutive equations (e.g. ideal gas law) or obtain physical
properties from tables. For First Law balances involving ideal gases with
constant heat capacities, it is often useful to set the reference state for the
energy such that U = CVT and H = CPT.
6. Solve the equations using symbols, rather than numerical values, as far
along as it possible.
7. Substitute numerical quantities into the final formula to obtain the an-
swers.
Some examples of applications of this methodology in problems involving First
Law balances follow.
Example 2.7 Underground storage of carbon dioxide
CO2 injection into depleted oil and gas formations underground is a key element of
planned carbon capture and storage activities to mitigate buildup of greenhouse
gases in the atmosphere. Consider CO2 from a pipeline at 150 bar and 20 C being
injected into a formation at P = 20 bar. A concern is that CO2 cools upon depressuri-
zation, leading to formation of hydrates that could block the borehole. You are
asked to (a) calculate the temperature of CO2 after depressurization, (b) determine
to what temperature CO2 should be heated prior to injection so that the tempera-
ture after depressurization is no less than 10 C and (c) calculate the energy re-
quirement of the heating process per mole of CO2 injected.
We will assume that there are no heat
losses (or input) as the CO2 moves
down the injection pipe. In this exam-
ple, the properties of the CO2 for-
mation itself, an open system that is
not at steady state, are not of particu-
lar interest. Instead, the system of in-
terest is the injection pipe and the vi-
cinity of the entrance to the reservoir,
where the pressure drop of the inject-
ed gas takes place. The selection of the
(open) system of interest is shown
with a dashed line the figure.
Figure
2.14.
Schematic
for
Example
2.7
2.6 Problem-Solving Strategies and Additional Examples
29
(a) A steady-state, open system First Law balance gives:
dU
1 NH
2 H2 = H1
= 0 = Q + W + NH
! dt
where
N
is
the
flow
rate
of
CO2 .
The
enthalpy
for
P1 =
150
bar,
T1 =
20
C
can
be
found
from
the
NIST
chemistry
WebBook,
using
either
isothermal
or
isobaric
table
lookup
we
obtain
H1
=
10.926
kJ/mol
.
Now
we
need
to
find
the
temperature
for
which
H2
=
10.926
kJ/mol
at
P2
=
20
bar.
This
calls
for
an
isobaric
property
calculation.
This
enthalpy
is
found
to
corre-
spond
to
a
mixture
of
saturated
liquid
and
gas
at:
T2
=
19.5
C
This
is
a
temperature
at
which
hydrates
can
indeed
form.
(b)
The
enthalpy
at
T2 =
10
C,
P2 =
20
bar
is
obtained
as
H2 =
20.722
kJ/mol.
An
isobaric
calculation
at
P1 =
150
bar
gives
the
desired
initial
temperature
to
match
this
specific
enthalpy
as:
T1
=
104.4
C
(c)
The
updated
steady-state,
open
system
balance
on
the
pipeline
now
reads:
dU
1 H 2 ) Q = H 2 H 1
= 0 = Q + W + N(H
N
! dt
Q
= !(!20.722 !10.926)!kJ/mol!=!+!9.796!kJ/mol
(heating
required)
!N
Example
2.8
Air
rifle
A
compressed-air
rifle
operates
as
follows.
The
air
chamber
of
volume
V0
=
10
cm3
initially
contains
air
at
P0
=
3
bar
(absolute)
and
0
=
25
C.
Releasing
the
trigger
ac-
celerates
a
projectile
of
mass
m
=
2
g
through
the
horizontal
barrel
of
cross
section
A
=
0.3
cm2
and
length
l
=
50
cm.
(a)
Calculate
the
velocity
of
the
projectile
as
it
exits
the
barrel.
(b)
After
exiting
the
barrel,
the
projectile
hits
a
wall
and
stops.
Calculate
its
tem-
perature
after
the
impact.
Air
can
be
considered
an
ideal
gas
with
CV
=
20.8
J/(mol
K).
The
projectile
has
heat
capacity
CV
=
0.126
J/(g
K).
The
atmosphere
is
at
Patm
=
1
bar.
30
Chapter 2. Work, Heat, and Energy
Referring
to
Fig.
2.15,
the
initial
volume
of
the
compressed
gas
is
V0
=
10
cm3;
the
final
volume
just
before
the
projectile
has
exited
the
barrel
is
Vf = (10 + 0.3 50)!cm 3 = 25!cm 3 .
!
(a)
We
will
assume
that
there
is
no
heat
transfer
to
or
from
the
gas
as
it
expands
(adiabatic
system)
and
that
there
is
no
friction
with
the
walls
of
the
barrel.
We
can
obtain
the
final
temperature
from
the
relationship
between
temperature
and
volume
for
adiabatic
expansion
of
an
ideal
gas,
Eq.
2.30:
Tf V
f
=
T V
0
! 0
R
CV
25
Tf = 298!K!
10
8.314
20.8
= !206.6!K
The
work
performed
by
the
gas
can
be
obtained
from
a
First
Law
balance
between
initial
and
final
states,
U = Q +W W = U = NCV T =
P0V0
RT0
CV T
3105 !Pa!! !10106 !m3
J
!20.8!
(206.6 298)!K!=! 2.30!J
J
mol!K
8.314!
!298!K
mol!K
!
This
work
is
negative
because
it
was
produced
by
the
gas
on
the
environment.
The
energy
goes
into
increasing
the
kinetic
energy
of
the
projectile
and
in
pushing
back
the
atmosphere
to
understand
why
this
term
should
be
taken
into
account,
consider
a
firing
of
the
rifle
in
space;
the
net
force
on
the
projectile
would
have
been
greater
in
this
case,
thus
resulting
in
greater
final
velocity
of
the
projectile.
On
earth,
a
First
Law
balance
for
the
system
projectile
+
atmosphere
gives:
Figure
2.15.
Schematic
for
Example
2.8
31
2.6 Problem-Solving Strategies and Additional Examples
2!(W + Patm V )
1
E = Q +W = mu2 Patm V u =
2
m
=
!
2!(2.30!J! !10 !Pa! !1510 !m )
m
!=!28.3!
0.002!kg
s
For
the
system
projectile
+
atmosphere,
W
=
+
2.30
J,
as
this
represents
work
input.
(b)
We
will
assume
here
that
all
the
kinetic
energy
of
the
projectile
is
turned
into
heat
when
the
projectile
hits
the
wall.
Then
1
0.002!kg! !28.32 !m 2 /s 2
U = mCV T = mu2 T =
= 3.18!K
2
2
2!g!
0.126!J/(g!K)
!
Example
2.9
Turbine
power
generation
A
turbine
is
used
to
generate
power
from
low-pressure
steam
obtained
from
a
so-
lar
collector
at
in
=
200
C
and
Pin
=
3
bar
at
a
rate
of
0.12
kg/s.
The
outlet
of
the
turbine
is
saturated
vapor
at
out
=
140
C.
If
the
heat
losses
of
the
turbine
are
3
kW,
obtain
the
power
(in
kW)
that
the
turbine
generates.
Steady-state,
First
Law
balance
on
the
turbine
gives
dU
= Q + W + N(H in H out ) W = Q + N(H
out H in )
dt
!
Properties are obtained from the NIST WebBook as:
(saturated steam, 140 C) = 2733.4 kJ/kg
!H out
(200 C, 3 bar) = 2865.9 kJ/kg
!H in
kg
kJ
W = +3!kW + 0.12 (2733.4! !2865.9) = !12.9!kW
s
kg
!
Chapter 3. Reversibility and Entropy
We have seen in the previous chapter that systems can move from an initial equi-
librium state to a different final equilibrium state by exchanging heat and work
with their surroundings. A new state can also be generated when a constraint, such
as a barrier to mass or heat flow, is released. The concept of the directionality of
change, however, has not yet been addressed. Everyday experience suggests that
some kinds of processes, even though in full compliance with the First Law, never
occur spontaneously. Examples of spontaneous processes are given in the table be-
low; the reverse processes have never been observed in isolated systems.
Initial state
Final state
Inflated helium balloon
Helium balloon deflated
Two metal blocks in contact
at different temperatures
Two metal blocks in contact
at the same temperature
Sugar crystals at the bottom
of a glass of pure water
Sugar dissolved in the water
Dispersed droplets of oil in water
Oil floating as separate layer on top of
the water
The
last
two
of
these
examples
(sugar
dissolving
in
water,
but
oil
separating
from
water)
illustrate
that
it
is
not
always
obvious
which
direction
(mixing
or
de-
mixing)
corresponds
to
spontaneous
change.
However,
once
the
spontaneous
change
direction
has
been
established,
it
is
impossible
to
convert
a
system
in
the
final
state
column
back
to
the
initial
state
without
the
action
of
an
external
agent.
For
example,
sugar
crystals
will
not
precipitate
out
of
solution
unless
the
water
is
evaporated.
Material
from
Essential
Thermodynamics,
Drios
Press
(2011),
A.Z.
Panagiotopoulos
36
3.1 Reversibility
37
3.1
Reversibility
Clearly,
all
processes
observed
naturally
are
spontaneous
and
thus
irreversi-
ble.
Spontaneous
changes
define
the
direction
of
the
arrow
of
time
and
are
rigor-
ously
analyzed
through
the
Second
Law
of
thermodynamics,
which
is
formally
in-
troduced
in
3.2.
By
eliminating
friction
and
all
gradients
of
temperature
or
pres-
sure,
and
by
performing
changes
at
an
infinitesimally
slow
rate,
one
can
approxi-
mate
an
idealized
reversible
process.
A
process
in
a
system
going
from
an
initial
to
a
final
state
is
formally
defined
as
reversible
if
the
system
can
be
brought
back
to
its
initial
state
from
the
final
state
with
no
change
to
any
part
of
the
universe.
Re-
versibility
is
an
idealized
abstraction
that
can
be
approximated
in
real
processes,
if
care
is
taken
to
avoid
friction,
internal
or
external
gradients
of
temperature,
pres-
sure,
or
composition,
as
well
as
any
conversion
of
mechanical
work
into
heat.
In
reversible
processes,
forces
across
any
boundary
exact-
ly
balance
at
all
times.
Pro-
cesses
take
an
infinitely
long
time
to
complete,
be-
cause
rates
of
heat
transfer,
macroscopic
flow
of
mate-
rial,
and
component
diffu-
sion
are,
respectively,
pro-
Figure
3.1
Rupture
of
a
partition
in
an
insulated
con-
portional
to
gradients
of
tainer.
temperature,
pressure,
and
chemical
potentials
(the
latter
defined
in
Chapter
5).
Consider
the
process
schematically
illustrated
in
Fig.
3.1.
A
thin
wall
separates
two
sides
of
an
insulated
container
in
the
initial
state
(A);
the
left-hand
side
con-
tains
a
gas
and
the
right-hand
side
is
evacuated.
The
wall
is
removed
and
the
gas
expands
to
fill
all
available
space.
After
some
time,
the
gas
reaches
a
new
equilibri-
um
state,
(B).
A
First
Law
balance
on
the
total
contents
of
the
container
gives
U
=
Q
/
+
W
/
=
0.
If
the
gas
is
ideal,
its
energy
depends
only
on
the
temperature,
and
T
=
0,
but
this
is
not
an
essential
characteristic
of
the
process;
for
real
gases,
the
tem-
perature
of
state
B
can
be
lower
or
higher
than
that
of
state
A.
We
can
return
the
gas
to
its
original
state
A
by
using
a
piston
to
compress
it
back
to
the
original
volume,
as
shown
in
Fig.
3.2.
For
this
reverse
process,
the
change
of
en-
Figure
3.2
Reversing
the
effect
of
expansion.
ergy
of
the
gas
is
the
same
38
Chapter 3. Reversibility and Entropy
as
previously,
U
=
0
=
Q
+
W
Q
=
W
;
since
we
clearly
need
to
provide
work
to
compress
the
gas,
the
re-
verse
process
is
only
pos-
sible
if
we
remove
heat
from
the
system.
If
some-
how
we
could
convert
all
Figure
3.3
Reversible
expansion.
of
the
heat
removed
into
work,
then
the
overall
pro-
cess
has
no
net
effect
on
the
environment
and
would
have
been
reversible
accord-
ing
to
the
definition
above.
However,
countless
experiments
have
shown
that
complete
conversion
of
heat
into
work
is
impossible;
we
thus
conclude
that
the
original
process
!A B
was
indeed
irreversible.
We
now
consider
a
different
process
(Fig.
3.3)
starting
from
the
same
initial
state
A
as
previously,
with
final
state
C
of
the
same
volume
as
B.
However,
we
now
perform
an
adiabatic
process
(!Q = 0 ),
using
a
well-lubricated,
frictionless
piston
to
obtain
work
from
the
expanding
gas.
The
amount
of
work
produced
by
the
gas
can
be
calculated
from:
V
WAC = C P dV
VA
!
Gases
generally
cool
as
they
expand
adiabatically,
so
that
the
final
tempera-
ture
will
be
TC
<
TA.
For
the
reverse
process
of
adiabatic
compression,
we
need
to
input
work,
which
in
the
absence
of
friction
will
be
equal
in
magnitude
and
oppo-
site
in
sign,
WCA
=
WA C.
Thus,
there
is
no
overall
net
effect
of
the
process
ACA
on
either
system
or
its
environment.
This
process
is
reversible.
3.2 The Second Law of Thermodynamics
The Second Law of thermodynamics places the concept of spontaneous changes on
a rigorous, quantitative basis. It allows for precise predictions about which trans-
formations can or cannot occur naturally in the absence of external driving
forces. It represents, in a highly compact fashion, the accumulated scientific
knowledge gained from countless experiments in carefully constructed systems.
While it cannot be proven from first principles and has to be postulated, it repre-
sents one of the most basic and well-established tenets of modern science.
This,
however,
is
not
true
for
all
substances.
3.1 Reversibility
39
The
Second
Law
can
be
stated
in
a
variety
of
equivalent
ways;
in
the
following,
we
use
a
traditional
approach
that
expresses
it
in
terms
of
heat
fluxes
around
heat
engines.
Heat
engines
are
devices
that
exchange
heat
and
work
with
their
envi-
ronment
in
processes
that
leave
no
net
effects
on
the
engines.
An
internal
combus-
tion
engine
undergoing
a
full
cycle
of
compression-ignition-expansion
is
a
classic
example
of
such
a
device.
However,
living
cells,
photovoltaic
arrays,
and
distilla-
tion
columns
can
also
be
considered
as
heat
engines,
provided
that
no
net
changes
accumulate
within
the
devices
over
the
period
of
interest.
Analysis
of
heat
engines
becomes
particularly
simple
if
they
exchange
heat
with
heat
reservoirs
large
enough
so
that
their
temperature
can
be
considered
con-
stant,
irrespective
of
the
amount
of
heat
exchanged
with
the
engines.
In
essence,
this
implies
that
the
amount
of
energy
exchanged
is
small
relative
to
the
total
heat
capacity
of
the
reservoirs.
Practical
approximations
of
heat
reservoirs
include
the
atmosphere,
rivers,
or
the
sea,
when
considered
over
short
enough
periods
of
time
so
that
their
temperature
does
not
vary
significantly.
Electrical
power
plants
are
usually
built
near
large
bodies
of
water
precisely
so
that
they
can
use
them
as
heat
reservoirs.
We
will
assume
that
heat
reservoirs
do
not
perform,
or
require,
any
work
in
the
process
of
interest.
The
Kelvin-Planck
postulate
(statement
of
the
Second
Law)
is
that:
It
is
not
possible
for
a
heat
engine
that
interacts
with
a
single
heat
reservoir
to
convert
all
the
heat
transferred
from
the
reservoir
into
work.
In
other
words,
heat
cannot
be
completely
converted
into
work.
However,
the
re-
verse
of
this
process,
converting
work
entirely
into
heat,
is
perfectly
possible.
Fig.
3.4
depicts
a
process
that
is
impossible
according
to
the
Kelvin-Planck
statement
of
the
Second
Law,
assum-
ing
that
the
signs
of
the
flows
of
heat
Q
and
work
W
are
as
indicated
by
the
arrows.
If
|Q|
=
|W|,
such
a
device
does
not
violate
the
First
Law,
as
a
trivial
balance
around
the
en-
gine
demonstrates.
No
such
device
has
ever
been
con- Figure
3.4
An
impossible
process.
structed,
despite
efforts
by
many
people
since
the
dawn
of
technological
civilization.
The
practical
value
of
being
able
to
extract
useful
en-
ergy
in
the
form
of
electricity
or
mechanical
work
by
cooling
the
sea
or
the
atmos-
phere
would
be
enormous;
fossil
fuel
use
could
then
be
reduced
to
zero
and
a
prac-
tically
inexhaustible
supply
of
energy
would
become
available
to
humanity.
Na-
ture,
however,
does
not
permit
such
a
device
ever
to
function.
Even
in
microscopic
systems,
where
spontaneous
fluctuations
occur
that
seem
to
result
in
mechanical
work
being
produced
at
the
expense
of
the
thermal
energy
of
a
reservoir
(e.g.
Brownian
motion
of
small
particles),
the
Second
Law
is
found
to
hold
when
the
av-
erage
behavior
of
a
system
is
determined
over
long
times.
Devices
that
violate
the
Kelvin-Planck
statement
of
the
Second
Law
40
Chapter 3. Reversibility and Entropy
First
or
Second
Laws
which
thus
can
never
be
constructed
are
called
perpetual
motion
machines
of
the
first
or
second
kind,
respectively.
3.3 Carnot Engines and Absolute Temperature
The seemingly simple statement of
the impossibility of certain process-
es made by the Second Law has
enormous consequences on the be-
havior of natural and engineered
systems. To start analyzing its con-
sequences, consider the behavior of
heat engines operating between two
heat reservoirs of different thermo-
metric temperatures, H and C , as
shown in Fig. 3.5. We will assume Figure 3.5 Thermal engines operating between
that the top reservoir is at a higher two heat reservoirs. Left side (engine A) corre-
sponds to power production and right side
thermometric temperature, H > C.
(engine B) corresponds to refrigeration.
Thermometric temperature in phys-
ical systems is observed to increase as energy flows into them.
On the left side of Fig. 3.5, engine A removes heat from the hot reservoir and
dumps some of it to the cold reservoir, while producing some work. Engine B on
the right panel does the reverse, removing heat from the cold reservoir and dump-
ing it to the hot reservoir through the consumption (input) of work. Nothing in the
First or Second Laws of thermodynamics prohibits any of these processes; practi-
cal examples of such systems, in the form of thermal power generation systems
and refrigeration units are ubiquitous. Nevertheless, there must be some con-
straints in the magnitudes of the work and heat streams from these engines, be-
cause otherwise a combination of them could give rise to a perpetual motion ma-
chine. To see why this is the case, consider using the work output of a machine of
type A to provide the energy input to a machine of type B. Without loss of generali-
ty, we can assume that:
Q = QCB
! CA
This
is
the
case
since
we
can
always
adjust
the
size
of
the
heat
engine,
or
how
many
engine
cycles
are
performed
per
unit
of
time.
First
Law
balance
around
the
system
composed
of
the
engines
A+B
then
gives
the
net
work
input
as:
The
net
heat
input
is:
W = WB WA
!
41
3.3 Carnot Engines and Absolute Temperature
Q = QHA QHB = W
!
If
W
<
0,
then
Q > 0 QHA > QHB
!
Net
work
is
being
produced
by
the
two
engines
by
removing
heat
from
the
hot
res-
ervoir.
This
is
equivalent
to
a
perpetual
motion
machine
of
the
second
kind,
violat-
ing
the
Kelvin-Plank
postulate,
so
it
is
an
impossibility.
Therefore,
we
must
have:
W 0 Q 0 QHA QHB
!
for
any
two
heat
engines
operating
between
two
specific
temperatures
H
and
C
.
The
arguments
of
the
previous
paragraph
also
apply
to
the
special
case
of
re-
versible
heat
engines,
known
as
Carnot
engines.
For
reversible
engines
we
can
simply
switch
the
role
of
A
and
B
at
will,
which
gives:
rev
rev
rev
rev
QHA
QHB
!!and!! QHB
QHA
rev
rev
QHA
= QHB
This
implies
that
for
any
reversible
heat
engine
that
operates
between
H
and
C
the
ratio
of
heat
fluxes
is
a
universal
function
of
the
two
temperatures:
QHrev
QCrev
= f (H ,C )
!
The
specific
form
of
the
function
in
Eq.
3.1
can
now
be
determined,
by
consider-
ing
a
cascade
of
heat
engines
operating
be-
tween
a
high
temperature
H,
an
interme-
diate
temperature
M
and
a
low
tempera-
ture
C,
as
shown
in
Fig.
3.6.
On
the
left
side,
two
Carnot
engines,
1
and
2,
are
used
between
H and
M
and
between
M
and
C
,
respectively.
On
the
right
side,
a
single
engine
3
operates
directly
between
H
and
C.
We
can
adjust
the
heat
flows
for
en-
gines
1
and
2
so
that
the
same
amount
of
heat
is
taken
and
removed
from
the
in-
termediate-temperature
reservoir
at
M,
and
we
can
also
adjust
engine
3
so
that
the
same
amount
of
heat
is
removed
from
the
hot
reservoir
as
for
engine
1.
Since
these
are
Carnot
(reversible)
engines,
we
must
have
that
|W1|
+
|W2|
=
|W3|.
Therefore,
the
(3.1)
Figure
3.6
Thermal
engines
operating
between
three
heat
reservoirs.
42
Chapter 3. Reversibility and Entropy
same
amount
of
heat
is
dumped
to
the
cold
reservoir
on
the
two
sides
of
the
cas-
cade:
QC = QC
QM / f (M ,C ) = QH / f (H ,C )
QH
QH
f (H ,M ) f (M ,C ) = f (H ,C )
f (M ,C ) f (H ,M ) f (H ,C )
!
Since
this
relationship
is
valid
for
any
temperatures
H,
M,
and
C,
the
func-
tional
form
of
f
must
be:
!
g(H )
f (H ,C ) =
g(C )
(3.2)
The
function
g()
is
universal
for
all
Carnot
engines.
Its
form
can
be
obtained
from
analysis
of
the
operation
of
any
reversible
heat
engine.
When
this
analysis
is
performed
for
engines
that
have
ideal
gases
as
working
fluids
(see
4.1),
we
ob-
tain
a
particularly
simple
relationship:
g()
=
T,
where
T
is
the
temperature
that
was
defined
through
the
ideal-gas
equation
of
state
in
2.4.
This
remarkable
prop-
erty
provides
a
connection
between
the
efficiency
of
reversible
engines
and
the
ideal-gas
temperature
scale.
The
absolute
temperature
scale
T
is
of
fundamental
importance
in
thermodynamics.
Given
that
f (H,C)
=
TH/TC,
we
can
now
show
that
reversible
heat
flows
occur-
ring
to
and
from
constant-temperature
reservoirs
are
linked
in
a
particularly
sim-
ple
way.
The
heat
flows
for
the
cold
and
hot
reservoirs
are
of
opposite
signs,
so
Eq.
3.1
can
be
expressed
as:
QHrev
QCrev
TH
TC
QHrev
TH
QCrev
TC
= 0
(3.3)
The
efficiency
of
a
Carnot
engine
that
draws
heat
from
a
hot
reservoir
and
produces
work
(left
side
of
Fig.
3.5)
is
defined
as
the
fraction
of
heat
removed
from
the
hot
reservoir
that
is
obtained
from
the
device
as
work.
From
a
First
Law
bal-
ance
around
a
reversible
engine,
we
have:
Carnot
Engine
efficiency
T
QHrev + QCrev +W = 0 QHrev C QHrev = W
TH
!
rev
!
W
QHrev
TH TC
TH
(3.4)
43
3.3 Carnot Engines and Absolute Temperature
The
efficiency
is
between
0
and
1,
the
latter
value
being
a
theoretical
limit
that
can
only
be
asymptotically
approached
if
TC0
or
TH+.
Real
engines,
of
course,
operate
with
efficiencies
lower
than
those
of
reversible
engines,
since
oth-
erwise
an
irreversible
engine
could
be
combined
with
a
Carnot
engine
operating
in
reverse,
resulting
in
a
violation
of
the
Second
Law.
The
performance
of
real
en-
gines
operating
between
reservoirs
of
given
temperatures
is
not
the
same
for
all
irreversible
engines
it
varies
(sometimes
greatly),
depending
on
their
design
and
internal
losses.
The
reversible
engine
efficiency
provides
a
common
upper
limit
for
all
real
engines.
Example
3.1
Efficiency
of
geothermal
power
production
In
many
parts
of
the
world,
especially
near
edges
of
tectonic
plates,
relatively
high
temperatures
can
be
reached
by
drilling
to
moderate
depths.
Taking
advantage
of
these
high
temperatures
has
been
proposed
as
one
possible
technology
for
energy
generation
without
production
of
greenhouse
gases.
Assuming
that
heat
can
be
withdrawn
from
hot
rock
at
H
=
200
C
and
that
cooling
is
available
at
C
=
50
C,
what
is
the
maximum
possible
fraction
of
heat
removed
that
can
be
converted
to
electricity?
The
maximum
possible
fraction
of
heat
conversion
into
work
takes
place
using
a
reversible
Carnot
engine,
which
has
efficiency:
T T
200 50
rev = H C =
= 32%
TH
200 + 273
!
Note
that
only
absolute
temperatures
[in
K]
can
be
used
in
the
expression
for
;
however,
the
difference
between
two
temperatures
in
C
is
the
same
as
the
corre-
sponding
absolute
temperature
difference
in
K,
so
the
thermometric
temperature
values
can
be
used
in
the
numerator.
For
engines
operating
as
refrigeration
cycles
(right
side
of
Fig.
3.5,
p.
40),
with
heat
removed
from
the
cold
reservoir
through
the
net
input
of
work,
a
measure
of
performance
different
from
is
appropriate.
In
such
cases,
we
are
interested
in
the
amount
of
heat
removed
from
the
cold
side
(the
interior
of
the
refrigerator,
or
the
inside
of
a
building
in
the
case
of
air
conditioning)
per
unit
of
work
input.
The
coefficient
of
performance,
,
is:
! = !
!
QC
W
QC
QC QH
rev !=
QCrev
QCrev
T
+ H QCrev
TC
TC
TH TC
(3.5)
Another possibility is the operation of a cycle as a heat pump, to maintain high-
er temperatures inside a building in the winter, by withdrawing heat from the cold
44
Chapter 3. Reversibility and Entropy
outside
air.
In
this
case,
the
coefficient
of
performance
is
defined
as
the
ratio
of
heat
output
(into
the
building)
over
the
work
input:
!=!
QH
W
QH
QC QH
rev =
QHrev
T
C QHrev + QHrev
TH
TH
TH TC
(3.6)
The
two
coefficients
of
performance
defined
in
Eqs.
3.5
and
3.6
can
be
greater
than
1,
if
the
difference
between
hot
and
cold
temperatures
is
smaller
than
the
ab-
solute
value
of
the
cold
or
hot
temperature,
respectively.
Practical
power
and
re-
frigeration
cycles
are
discussed
in
Chapter
4.
Example
3.2
Air
conditioner
theoretical
efficiency
Calculate
the
maximum
possible
coefficient
of
performance
for
an
air
conditioning
unit
operating
between
an
indoor
temperature
of
68
F
and
outdoor
temperature
of
104
F.
We
first
need
to
convert
thermometric
to
absolute
temperatures:
C
=
68
F
C
=
293
K;
C
=
104
F
TH
=
313
K.
The
maximum
possible
coefficient
of
performance
is:
rev
!=!
QCrev
W
TC
TH TC
293
= 14.6
313 293
3.4 Entropy Changes in Closed Systems
We showed in the previous section that reversible heat flows occurring to and
from constant-temperature reservoirs satisfy the condition QHrev /TH + QCrev /TC = 0 ;
!
this in turn suggests that a heat-related property can be defined that is conserved
for a system undergoing a reversible process that starts and ends at the same state
point. This new property is called the entropy S. For a reversible process in closed
systems for which all exchange of heat is done at a single temperature T, the en-
tropy change S is defined as:
Q rev
S
(3.7)
T
!
For
a
differential
change
of
state
during
a
reversible
process,
the
temperature
of
the
system
is
effectively
constant,
so
we
can
write:
Q rev
dS =
T
!
(3.8)
3.4 Entropy Changes in Closed Systems
45
The
overall
entropy
change
for
a
general
process
that
involves
temperature
changes
along
its
path
is:
definition
of
entropy
rev
B Q
S = SB S A =
A T
!
(3.9)
A reversible process can always be constructed between any two equilibrium
states of a system, provided that external reservoirs are available to exchange heat
and work as needed. Thus, Eq. 3.9, along with a choice of a reference state for
which S = 0, in principle provides a way to generate values for the entropy of any
equilibrium state of a thermodynamic system. As will soon become apparent, Eq.
3.9 is not usually a practical approach to obtain actual entropy changes. Thermo-
dynamic relationships incorporating volumetric and heat capacity data, to be dis-
cussed later in the present section and in 7.3, are more commonly used for en-
tropy change calculations.
A key property of the entropy is that it always increases for spontaneous pro-
cesses in closed, isolated systems. This property is often used as an alternative
statement of the Second Law
here, we simply derive it from the
Kelvin-Planck postulate as follows.
Consider an irreversible, adiabatic
process taking place between ini-
tial state A and final state D, along
the dashed-line path of Fig. 3.7.
Curves AB and CD represent re-
versible adiabatic paths from the
initial and final state respectively,
and curve BC is an isothermal path
at temperature T. For the reversible
Figure 3.7 Pressure-volume relationship for ir-
process DCBA that brings the sys-
reversible process (dotted line) and adiabatic-
tem from state D back to the initial
isothermal-adiabatic path.
state A, all heat transfer takes place
along the isothermal step BC, so that:
rev
rev
rev
U UD = QDA
+WDA
= T(S A SD )+WDA
! A
For the irreversible process AD
irr
U U A = WAD
! D
Adding
these
two
expressions
we
obtain
rev
irr
T(S S )+WDA
+WAD
= 0
! A D
46
Chapter 3. Reversibility and Entropy
irr
rev
rev
+WDA
< 0 ,
then
T(S A SD ) = QDA
> 0 ;
the
overall
cycle
has
received
If
WAD
!
!
!
heat
input
from
a
single
reservoir
at
T
and
has
produced
net
work.
This
is
a
viola-
tion
of
the
Kelvin-Planck
postulate
and
therefore
impossible.
The
case
rev
W irr +WDA
= 0 implies
that
T(S A SD ) = Q rev = 0 and
the
final
state
could
be
re-
! AD
!
turned
to
the
initial
state
with
no
net
effect
on
the
environment;
this
contradicts
the
original
statement
that
the
process
AD
was
irreversible.
Therefore,
we
must
have,
for
any
irreversible
process
in
an
isolated
closed
system
from
state
AD,
T(S A SD ) < 0 ,
or
equivalently:
S > S [for irreversible process A D]
!D A
(3.10)
irr
rev
irr
rev
+WDA
> 0 WAD
> WDA
In
addition,
we
must
have
WAD
.
By
changing
the
!
direction
of
the
reversible
process
from
DA
to
AD
we
then
obtain:
rev
W irr > WAD
!!
! AD
(3.11)
Since
the
universe
can
be
considered
a
closed
isolated
system,
Eq.
3.10
is
the
mathematical
equivalent
of
the
Clausius
statement
of
the
Second
Law,
the
entropy
of
the
universe
tends
to
a
maximum.
Eq.
3.11
states
that
the
algebraic
work
amount
is
always
greater
for
an
irreversible
process.
If
the
reversible
process
re-
rev
!>!0! )
then
the
irreversible
process
requires
more
work
input;
quires
work
( WAD
!
rev
!<!0! ),
Eq.
3.11
suggests
that
the
ir-
if
the
reversible
process
produces
work
( WAD
!
reversible
process
will
produce
less
work
it
may
even
require
work
input!
Re-
versible
processes
are
the
best
of
all
possible
processes
in
achieving
a
given
task
with
the
least
expenditure
of
useful
work
and
in
extracting
the
maximum
possible
amount
of
useful
work
out
of
a
given
change
of
state.
Entropy
has
been
just
derived
from
analysis
of
heat
and
work
flows
in
reversi-
ble
heat
engines;
however,
once
it
has
been
established
that
entropy
is
a
proper
thermodynamic
function,
it
can
be
expressed
for
any
equilibrium
state
in
terms
of
any
convenient
thermodynamic
variables.
This
provides
the
starting
point
for
de-
velopment
of
thermodynamic
identities
through
the
formal
approach
described
in
Chapter
5.
Let
us
take
a
look
again
at
the
differential
form
of
the
First
Law
of
ther-
modynamics,
written
for
a
closed
system
undergoing
a
reversible
process:
rev
rev
!dU = Q + W
(3.12)
A differential change of state does not substantially change the temperature of
the system, so we can set Q rev = TdS . Since the pressure also remains constant
!
(within a differential amount), the work performed by the environment on the sys-
tem can be expressed as !W rev = PdV . Combining the expressions for Q rev and
!
rev
!W we get the fundamental equation of thermodynamics, which provides a syn-
thesis of the First and Second Laws:
47
3.4 Entropy Changes in Closed Systems
!dU = TdS PdV
(3.13)
Eq.
3.13
has
only
proper
thermodynamic
state
functions
on
both
sides,
contain-
ing
no
inexact
differentials
involving
heat
or
work
amounts.
Changes
in
state
func-
tions
are
independent
of
the
path,
so
the
equation
is
valid
for
all
processes,
re-
versible
or
irreversible.
In
irreversible
processes,
the
first
term
(TdS )
is
not
equal
to
the
amount
of
heat,
and
the
second
term,
(PdV )
does
not
equal
the
amount
of
work;
but
their
sum
still
gives
the
change
in
system
energy.
Example
3.3
Entropy
change
for
an
ideal
gas
10
mol
of
an
ideal
gas
with
CV
=
20.8
J/mol
at
T0
=
300
K
and
P0
=
0.3
MPa
occupy
the
left
half
of
an
insulated
vessel,
as
shown
in
Fig.
3.8.
The
other
half
is
evacuated.
At
time
t
=
0,
a
1
kW
electrical
heating
element
is
turned
on.
After
30
s,
the
parti-
tion
dividing
the
vessel
ruptures
and
the
heating
element
is
turned
off.
Calculate
(a)
the
final
temperature
Tf
and
pressure
Pf
of
the
gas
in
the
vessel
and
(b)
the
en-
tropy
change
of
the
gas
during
this
process.
The
final
temperature
of
the
gas
can
be
obtained
from
a
First
Law
balance
on
the
contents
of
the
vessel:
U = NCV T = Q + W Tf = T0 +
Q
NCV
30!s! !1000!J/s
Tf = 300!K! +
= 444!K
10!mol! 20.8!J/(mol!K)
!
The final pressure is obtained from the ideal-gas law,
Pf Vf
RTf
P0V0
RT0
Pf = P0
V0Tf
Vf T0
444!K
Pf = 0.3!MPa
= !0.222!MPa
2! !300!K
!
The
entropy
change
cannot
be
directly
obtained
from
the
definition
(Eq.
3.9),
as
this
is
not
a
reversible
pro-
cess.
Instead,
it
can
be
obtained
from
Eq.
3.13:
Figure
3.8
Schematic
of
pro-
cess
for
Example
3.3.
1
P
dU = TdS PdV dS = dU + dV
T
T
!
NCV
C
T
V
NR
S
R
dS =
dT +
dV
= V dT + dV = CV ln f + Rln f
T
V
N
T
V
T0
V0
!
Valid
for
any
process
in
a
closed
system
48
Chapter 3. Reversibility and Entropy
S
444
J
J
= 20.8ln
+ 8.314ln2 !
= (8.16 +5.76)!
N
300
mol!K
mol!K
J
J
S = 10!mol! 13.9
S = 139!
mol!K
K
!
!
The
entropy
change
is,
of
course,
greater
than
zero
during
this
irreversible
pro-
cess.
Using
the
same
approach
as
in
Example
3.3,
it
is
easy
to
prove
that
the
general
relationship
for
the
entropy
change
of
an
ideal
gas
with
temperature-independent
heat
capacities
during
a
process
that
takes
it
from
(T0 ,P0 ,V 0 ) (Tf ,Pf ,V f ) is:
Entropy
change
for
an
ideal
gas
S
!
IG
= CV ln
Tf
T0
+ Rln
Vf
V0
= C P ln
Tf
T0
Rln
Pf
P0
(3.14)
Eq.
3.14
is
of
general
applicability
to
changes
of
state
for
ideal
gases
the
pro-
cess
does
not
have
to
be
reversible,
at
constant
temperature
or
at
constant
pres-
sure.
Example
3.4
Forging
A
common
process
in
metallurgy
is
to
immerse
a
red-hot
item
(e.g.
a
blade
being
forged)
in
water
to
harden
it.
Let
us
consider
a
process
in
which
a
1
kg
steel
blade
at
1
=
800
C
is
immersed
in
a
large
vessel
filled
with
water
at
2
=
25
C
and
imme-
diately
quenched.
Assuming
that
steel
has
heat
capacity
CV
=
460
J/(kg
K),
inde-
pendent
of
temperature,
calculate
the
entropy
changes
during
this
process
of
(a)
the
steel
blade,
(b)
the
water,
and
(c)
the
universe.
Assume
that
there
is
enough
water
in
the
vessel
so
that
its
temperature
does
not
increase
appreciably
during
the
process
of
immersion.
The
entropy
change
of
the
steel
blade
in
this
irreversible
process
can
be
calculated
from
Eq.
3.13,
ignoring
the
small
volume
change
of
the
solid
as
it
cools
down:
dU = TdS PdV
!
NC dT
1
dS = dU = V
T
T
C
T
Sblade = N V dT = NCV ln f
T
T0
!
J
273+ 25
J
Sblade = 1!kg! !460!
!ln
= 589!
kg!K 273+ 800
K
!
49
3.4 Entropy Changes in Closed Systems
Even
though
this
is
an
irreversible
process,
the
blades
entropy
goes
down
the
blade
is
not
an
isolated
system,
so
Eq.
3.10
does
not
apply.
For
the
water,
we
are
not
given
enough
information
to
calculate
its
temperature
change;
since
the
process
is
clearly
irreversible,
it
would
seem
that
we
cannot
use
the
definition
of
entropy
change
(Eq.
3.9)
however,
this
is
not
true!
Even
though
the
overall
process
is
irreversible
because
of
the
large
temperature
difference
be-
tween
the
blade
and
the
water,
we
can
devise
a
thought
experiment
in
which
the
heat
transfer
to
the
water
is
done
in
a
reversible
manner.
The
amount
of
heat
transferred
is:
J
Q = U blade = NCV T = 1!kg 460!
(25 800)!K!=!356!kJ
kg!K
!
The
entropy
change
for
the
water
in
a
reversible
process
with
the
same
Q
is:
Q rev
356!kJ
J
Swater =
=
= 1196!
T
(273+ 25)!K
K
!
The
entropy
change
of
the
universe
is:
J
J
Suniverse = Swater + Sblade = (589+1196) = +607
K
K
!
Once
again,
the
entropy
change
of
the
universe
is
positive,
as
it
should
be
for
an
overall
irreversible
process.
3.5 Entropy Changes in Open Systems
The entropy of an open system can change because of heat exchanged with the en-
vironment, because of flows into and out of the system and because of entropy
generation due to internal irreversibilities entropy is not a conserved quantity!
While entropy can be created, a process can never result in a net decrease in the
entropy of the universe; this would violate the Second Law of thermodynamics.
This condition translates into an inequality linking the entropy change of the sys-
tem Ssystem, the entropy change of the environment Senv, and the specific entro-
pies of entering and leaving streams:
Suniverse = Ssystem + Senv +
!
leaving
streams
Nout S out
entering
streams
Nin S in 0 (3.15)
Why
are
the
terms
for
the
entering
and
leaving
streams
in
Eq.
3.15
of
opposite
sign
relative
to
Eqs.
2.16
or
2.19
(p.
18),
the
First
Law
balance
for
open
systems?
50
Chapter 3. Reversibility and Entropy
Eqs.
2.16
and
2.19
are
written
from
the
point
of
view
of
the
system,
which
gains
the
streams
that
enter
and
loses
the
streams
that
leave.
By
contrast,
Eq.
3.15
rep-
resents
a
balance
for
the
entropy
of
the
universe.
For
the
universe,
streams
enter-
ing
the
system
are
lost
and
streams
exiting
the
system
are
gained.
A
similar
re-
lationship
can
also
be
written
as
a
differential
(rate
of
change):
Suniverse = Ssystem + Senv +
leaving
streams
N out S out
entering
streams
N in S in 0
(3.16)
For
reversible
processes,
Eqs.
3.15-3.16
become
equalities,
with
their
right-
hand
side
equal
to
zero.
For
other
special
cases,
specific
terms
can
be
set
to
zero,
significantly
simplifying
the
equations.
For
example,
the
term
Ssystem
is
zero
at
steady
state,
as
there
is
no
net
change
in
the
system.
Another
example
of
simplifi-
cation
is
for
an
adiabatic
process,
for
which
we
can
set
Senv =
0,
since
no
heat
flows
into
the
environment.
The
fact
that
entropy
generation
is
non-negative
for
all
feasible
processes
im-
poses
significant
constraints
on
the
amount
of
work
that
can
be
produced
from
(or
is
required
for)
specific
processes.
When
performing
a
thermodynamic
analysis,
a
typical
approach
is
to
apply
in
turn
a
First
Law
balance
followed
by
a
Second
Law
balance,
as
illustrated
in
the
examples
that
follow.
Example
3.5
Feasibility
of
a
process
An
inventor
is
proposing
a
black
box
device
for
producing
electrical
power
that
operates
on
a
stream
of
compressed
air
at
P1
=
4
bar,
T1
=
300
K.
The
input
stream
is
split
into
two
equal
flows
of
P2
=
P3
=
1
bar
at
T2
=
280
K
and
T3
=
260
K,
respectively.
The
claim
is
made
that
power
is
produced
at
a
steady-state
rate
of
W
=
3.4
kJ/mol
of
air
flowing
through
the
device.
Assuming
that
unlimited
heat
exchange
with
the
environment
at
Tenv
=
300
K
is
allowed,
you
are
asked
to
provide
an
analysis
of
the
thermodynamic
feasibility
of
such
a
device.
Assume
that
air
can
be
approximated
as
an
ideal
gas
with
CP
=
29.1
J
/(mol
K),
independent
of
temperature.
First,
we
apply
an
integral
First
Law
balance
on
this
open
system
over
the
pe-
riod
of
time
it
takes
for
1
mol
of
air
to
flow
through
the
device,
which
is
as-
sumed
to
be
at
steady
state.
The
flow
of
streams
2
and
3
is
half
the
flow
of
stream
1.
We
use
a
refer-
ence
state
for
the
enthalpy
Figure 3.9 Schematic of process for Example 3.5.
51
3.5 Entropy Changes in Open Systems
such that H = C PT .
U
= 0 = Q +W + NH 1
!steady!state
T +T
N
N
H 2 H 3 Q = W NC P T1 2 3
2
2
2
J
260+ 280
Q = (3400)!J! 1!mol!
!
!29.1!
300
K != 2527!J
mol!K
2
!
Since
the
heat
calculated
from
the
systems
point
of
view
is
positive,
heat
flows
from
the
environment
to
the
system
during
this
process.
We
must
now
check
whether
the
entropy
generation
rate
for
the
universe
is
non-negative.
We
use
Eq.
3.14
(p.
48)
to
calculate
the
entropy
change
of
the
ideal
gas
streams
and
take
into
account
that
the
flow
of
heat
into
the
environment
is
the
opposite
of
the
value
cal-
culated
from
First
Law
balance
on
the
system:
Suniverse =
Sdevice + Senv + Nout S out Nin S in =
leaving!
entering!
steady!state
!
= !
streams
streams
Q N
N
Q N
+ S 2 + S 3 N S1 =
+ (S 2 S 1 + S 3 S 1 )
Tenv 2
2
Tenv 2
TT
PP
Q N
+ C P ln 2 3 Rln 2 3 =
Tenv 2
T12
P12
2527!J 1!mol
J
260 280
J
1
!+!
29.1!
ln
8.314
ln 2 =
2
300!K
2
mol!K
mol!K 4
300
J
J
!!!!!!!!!!!!!!!!!!!!!!!!!!! 8.42 3.09!+11.53 ! = +0.02!
K
K
!
The
entropy
change
of
the
universe
is
positive,
so
operation
of
the
device
is
possi-
ble
as
described.
However,
since
the
entropy
change
is
small
relative
to
the
terms
of
which
it
consists,
the
device
is
operating
near
the
thermodynamic
limit
for
re-
versible
processes.
Example
3.6
Solar
power
generation
Solar
collectors
are
used
to
heat,
continuously
and
at
steady
state,
a
molten
salt
stream
from
200
C
to
600
C
at
a
flow
rate
of
12
kg/s.
The
hot
stream
generates
electrical
power
by
a
complex
process
and
then
returns
to
the
solar
collectors.
What
is
the
maximum
amount
of
power
that
can
be
produced?
Assume
that
the
environment
is
at
20
C.
The
heat
capacity
of
the
molten
salt
at
constant
pressure
is
CP
=
0.8
kJ/(kg
K),
independent
of
temperature.
52
Chapter 3. Reversibility and Entropy
Figure 3.10 Schematic of process for Example 3.6.
The
system
of
interest
is
the
power
plant
that
uses
the
salt
stream.
We
label
the
hot
stream
H
and
the
cold
one
C.
An
open-system
First
Law
balance
for
this
sys-
tem
gives:
dU
=0
dt
H HC )
= Q + W + N(H
steady!state
W = Q + NC P (TC TH )
!
The
maximum
amount
of
work
is
produced
in
a
reversible
process,
for
which
Suniverse = 0 =
Splant + Senv + Nout S out Nin S in =
leaving!
entering!
steady!state
streams
streams
T C
T
Q
ln C
+ N(S C SH ) Q = Tenv N C P dT = Tenv NC
P
T
Tenv
TH
H T
T
T ln H +T T
W = NC
P
env
C
H
TC
273+ 600
kg
kJ
W = 12 0.8
293!ln
+ 200 600 K!!= 2.12!MW
s
kg!K
273+ 200
!
The
power
is
negative
because
it
is
being
produced
by
the
power
plant.
Example 3.7 Analysis of a Steam Turbine
A power plant operates with supercritical steam at T1 = 900 K and P1 = 8 MPa as in-
put to a turbine. The steam exits at T2 = 430 K. Assuming that the turbine operates
53
3.5 Entropy Changes in Open Systems
reversibly and adiabatically, how much steam is required to generate power at a
rate of 1 MW?
Figure 3.11 Schematic of process for Example 3.7.
For
reversible
operation
of
the
steam
turbine,
we
must
have:
Suniverse = 0 = Sturbine + Senv + N(S 2 S 1 ) S 2 = S 1
reversible!process
steady!state
adiabatic
The
properties
of
steam
at
the
entrance
of
the
turbine
(T1
=
900
K
and
P1
=
8
MPa)
can
be
obtained
from
the
NIST
WebBook
as
follows:
kJ
kJ
H 1 = 3707! !!!!!S 1 = 7.095
kg
kg!K
!
We
now
need
to
find
the
pressure
for
which
S2
=
S1
=
7.095
kJ/(kg
K)
at
T2
=
430
K.
With
a
bit
of
trial-and-error
with
respect
to
the
pressure
range,
we
can
obtain
from
the
NIST
WebBook
P2
=
0.31
MPa,
H2
=
2775
kJ/kg.
A
First
Law
balance
on
the
tur-
bine
now
gives:
dU
= 0 = Q + W + N(H
1 H2 )
dT
!steady!state
W
103 !kJ/s
N =
=
H 2 H 1 (2775 3707)!!kJ/kg
!
Example
3.8
Work
for
evacuating
a
tank
kg
N = 1.073
s
!
A tank of volume V0 initially contains air at a certain temperature T0 and pressure
P0. The environment is also air at T0 and P0. Obtain general expressions for the
minimum amount of work required to evacuate or compress the tank to a pressure
P1 = P0 (a) isothermally and (b) adiabatically. Perform illustrative calculations for
= 0.1 and 10, and obtain the work required for complete evacuation ( = 0). Air
can be considered an ideal gas with CV = 5R/2.
54
Chapter 3. Reversibility and Entropy
This problem can be handled most conveniently
by transformation from an open to an equivalent
closed system. For the case of evacuation, we
would like to remove material from the tank of
volume V0 , initially at T0 and P0, to bring the pres-
sure down to P1. This process reduces the number
of moles in the tank from N0 to N1. Instead of ac-
complishing this by action of a pump, in the
Figure 3.12 Schematic of pro-
closed system process, a frictionless piston is set
cess for Example 3.8.
up as shown in Fig. 3.12, at a position within the
tank that contains an amount of material N1, has
volume V1, and is originally at T0 and P0. At the right-hand side of the piston there
is air at T0 and P0. We will pick V1 so that after the piston has moved from V1 to V0,
the final pressure within the tank is P1 = P0. This reversible process accomplishes
the same task as pumping the original tank (of volume V0) from P0 to P1. The mini-
mum work for evacuation is obtained as the work to move the piston rightward
from V1 to V0. A similar approach can give the minimum work required for com-
pression to a certain pressure, or the maximum work that can be obtained from a
cylinder of compressed air.
(a) Isothermal operation
For this case, from the ideal-gas equation-of-state, PV = NRT, the volume V1 is ob-
tained as:
V1
V
! 0
P1
P0
N1
N0
The
instantaneous
pressure
difference
across
the
piston
is
P
P0,
where
P
is
the
pressure
within
the
expanding
volume
on
the
left
side.
The
total
work
required
to
move
the
piston
to
the
right
is:
V
V0 N RT
1
V1
V1
W = 0 (P P0 )dV =
dV + P0(V0 V1 ) = N1RT ln
V1
V0
+ P0(V0 V1 )
V PV PV
P V
W N1
=
RT ln 1 + 0 0 0 1 = RT ln + RT 0 1 = RT ln + RT RT
N0 N0
V0 N0
N0
N1
W
W
=
= ln +1
RTN0 P0V0
For = 0.1, this equation gives W/(RTN0) = 0.670.
Because ln0 as 0, the expression has a simple interpretation at the limit of
complete evacuation:
55
3.5 Entropy Changes in Open Systems
W
W
=
= 1 W = P0V0
for
complete
evacuation.
!
N0RT P0V0
!
In
other
words,
the
minimum
work
for
complete
evacuation
is
the
work
to
push
back
the
atmosphere
by
a
volume
equal
to
the
original
tank
volume.
n
the
case
of
pressurization,
a
more
meaningful
measure
than
the
work
per
mole
initially
in
the
tank
is
the
work
per
mole
of
compressed
air,
obtained
by
dividing
the
full
expression
above
by
=
N1/N0,
W
W
1
=
= ln + 1
RTN1 P1V0
For
=
10,
this
equation
gives
W/(RTN1)
=
1.403.
The
work
required
is
positive
(work
input
to
the
system)
for
both
lowering
and
increasing
the
tank
pressure
to
a
value
different
than
atmospheric.
(b)
Adiabatic
operation
Here,
we
will
use
the
expressions
obtained
in
2.4
for
adiabatic
compression
and
expansion
of
an
ideal
gas.
Even
though
we
did
not
state
so
at
the
time,
adiabatic
operation
(Q
=
0)
at
internal
equilibrium
implies
also
a
reversible
operation,
since
the
entropy
change
of
the
universe
for
such
a
process
is
zero.
As
previously,
we
find
V1
so
that
P1
=
P0
after
expansion.
From
Eq.
2.31,
with
ini-
tial
volume
and
pressure
V1
and
P0
and
final
values
V0
and
P1
:
V
== 0
P
V1
! 0
P1
V1
V0
= 1/ =
CV /C P
The final temperature after expansion is obtained from Eq. 2.30:
1
P
R/C
= 1 = P
T
P
! 0 0
The
work
performed
on
the
gas
during
the
expansion
is,
from
Eq.
2.32:
T1
T
R/C
Ugas = Q +Wgas Wgas = N1CV T = N1CV (T1 T0 ) = N1T1CV 1 0 = N1T1CV 1 P
T1
!
The
net
work
required
is
the
sum
of
the
work
performed
on
the
gas
and
the
work
to
push
back
the
atmosphere,
56
Chapter 3. Reversibility and Entropy
W = Wgas + P0 (V0 V1 ) = N1T1CV 1
R/C P
) + P (V V )
0
N1T1CV
PV
PV
PV C
W
R/C P
=
1
+ 0 0 0 1 = 1 0 V
N0RT0
N0RT0
N0RT0 N0RT0
P0V0R
W
W
1/
=
=
+1 1/
RT0N0 P0V0
1
V
1 +1 1
V0
For
air,
=7/5=1.4,
so
that
for
=
0.1
we
obtain
W/(RTN0)
=
0.574.
A
little
less
work
is
required
for
adiabatic
evacuation
than
for
isothermal
one.
At
the
limit
0,
this
expression
also
gives
W
=
P0V0,
as
for
the
isothermal
case.
For
adiabatic
pressurization,
the
work
per
mole
of
compressed
air
is:
1/
W
W V0 W
W
11/ 1
1/
=
=
=
+11/
=
+ 1/ 1
RT N
1
RT N P V V1 RT0N0
1
! 0 1
! 0 1 1 0
For
=
10,
this
expression
gives
W
/
(RTN1)
=
1.520,
a
little
more
than
in
the
iso-
thermal
case.
3.6 Microscopic Origin of Entropy
By this point, we are familiar with the definition of entropy from a macroscopic
thermodynamic viewpoint as S = Qrev/T. But what is the origin of entropy in the
microscopic, molecular world? Qualitatively, we expect that irreversible processes
(which result in an increase in total entropy) also increase the degree of disorder
in a system. It turns out that a quantitative measure of disorder, specifically the
number of microscopic states available to a system at a given internal energy U
and for specified number of molecules (or moles) N and volume V, can be quantita-
tively linked to the entropy. A distinct microscopic state (microstate) is defined by
all microscopic degrees of freedom e.g. positions and velocities of molecules in a
gas. A set of microstates with specified common properties (e.g. number of parti-
cles N, volume V and energy U) defines an ensemble. The expression of S in terms of
microstates is provided by the famous 1872 entropy formula of Ludwig Boltz-
mann,
S = kB ln (N ,V ,U )
!
(3.17)
Boltzmanns entropy formula is the foundation of statistical mechanics, con-
necting macroscopic and microscopic points of view. It allows calculations of mac-
roscopic thermodynamic properties by determining properties of microscopic
3.6 Microscopic Origin of Entropy
57
configurations. The constant kB is called Boltzmanns constant; it is the ratio of the
ideal-gas constant to Avogadros number, or equivalently the gas constant on a per
molecule basis: kB = R/NA =1.3806510-23 J/K.
To illustrate the concept of counting microstates, we will use the simple sys-
tem shown in Fig. 3.13. In the general case, it consists of N slots, each containing a
ball that can be at energy levels 0, +1, +2, +3, , measured in units of kBT0, where
T0 is a reference temperature. The specific case of N = 10 and 3 energy levels is
shown in the figure. The concept of discrete energy levels arises very naturally in
quantum mechanics. For this simple system, we can count states by using the
combinatorial formula giving the number of ways we can pick M specific distin-
guishable objects out of N total objects:
N
N!
N (N 1)(N M +1)
(3.18)
= M!(N M)! =
12M
M
!
There
is
only
1
state
with
internal
energy
U
=
0.
States
with
U
=
1
have
one
ball
at
level
+1
and
the
others
at
level
0,
so
for
N
=
10,
there
are
10
such
states.
States
with
energy
U
=
2
may
have
1
ball
at
+2
and
the
others
at
0,
or
2
balls
at
+1,
so
there
are:
10
+10 = 55
2
!
such
states.
We
can
similarly
obtain
(3 )= 220;
(4 )= 715,
(5 )= 2002,
and
so
on.
Note
that
the
number
of
microstates
increases
rapidly
with
the
total
energy
of
the
system.
This
is
generally
the
case
for
most
systems.
Now
we
are
in
a
position
to
show
that
S
defined
microscopically
from
Eq.
3.17
has
the
two
key
properties
associated
with
the
entropy
of
classical
thermodynam-
ics:
1.
S is extensive: For two independent subsystems, A and B,
S = k ln( A+B ) = kB ln( A B ) = kB ln A + kB lnB
! A+B B
The reason is that each microstate of system A can be combined with a
microstate of system B to give a microstate of the combined system. This is
clearly true for the simple system illustrated on the previous page. However,
when mixing two
gases or liquids, we
only get the above
expression if we
assume that the
particles in the
systems
are Figure 3.13 A system of 10 spheres with 3 energy levels.
indistinguishable. If
58
Chapter 3. Reversibility and Entropy
particles are distinguishable, additional states are available to the combined
system resulting from the possibility of exchanging the labels of particles.
Although the indistinguishability of particles is really of quantum mechanical
origin, it was introduced ad hoc by Gibbs before the development of
quantum mechanics, in order to make entropy an extensive property.
2.
S is maximized at equilibrium: For a system with internal constraints (e.g.
internal rigid walls or barriers to energy transfer), the number of possible
microstates is always smaller than the number of microstates after the
constraints are removed.
S (N,V, U) > S (N,V, U; internal constraints)
To demonstrate this second property, consider the box with particles of the
example above, and think of any constraint to the system at a given total
energy (say U=+2). An example of a "constraint" would be to have that the
first five slots have exactly 1 unit of energy. The number of microstates in
this case is (5x5=25), less than the 55 states available to the unconstrained
system.
One clear distinction between macroscopic and microscopic definitions of en-
tropy is that the former is physically meaningful only as a difference of entropy be-
tween specified states, while the latter appears to provide a measure of absolute
entropy. This apparent discrepancy results from the inability of classical physics to
define uniquely when two nearby states (e.g. positions of a particle in free space
differing by a fraction of a nm) are sufficiently different to justify distinguishing
them from each other. Quantum mechanical methods, on the other hand, provide
precise ways to count states.
At low temperatures, the number of microstates available to any physical sys-
tem decreases rapidly. At the limit of absolute zero temperature, T 0 , most sys-
tems adopt a unique ground state for which = 1 S = kBln = 0. This is the basis
of the Third Law of thermodynamics postulated by Nerst in the early 1900s. The
NIST Chemistry WebBook lists absolute entropies for pure components and chem-
ical elements in the thermochemistry data section. However, using entropy values
calculated with respect to an arbitrary reference state gives the same results as
absolute entropies for heat and work amounts.
Example 3.9 Entropy of a lattice chain
A common model for polymers is the Flory lattice model, which represents chains
as self-avoiding random walks on a lattice (grid). Self-avoiding means that two
beads cannot occupy the same position on the lattice. When two non-bonded
beads occupy adjacent positions, they have energy of interaction equal to kBT0,
where T0 is a reference temperature. Obtain the number of microstates for a
two-dimensional square-lattice chain of 5 beads, as a function of the energy U of
the chain.
3.6 Microscopic Origin of Entropy
59
Fig.
3.14
shows
the
number
of
configurations
for
a
square-lattice
chain
of
5
beads.
Without
loss
of
generality,
we
have
fixed
the
configuration
of
the
first
two
beads
of
the
chain
to
be
in
the
horizontal
direction,
with
the
second
bead
to
the
right
of
the
first.
This
reduces
the
total
number
of
configurations
by
a
factor
of
4;
such
a
multi-
plicative
factor
simply
shifts
the
value
of
S
obtained
from
Eq.
3.17
by
a
constant
factor,
akin
to
the
reference
state
for
the
entropy.
The
last
bond
is
shown
in
multi-
ple
configurations
(arrows),
along
with
their
number
and
energy:
!2(1)
for
the
top
left
image
means
there
are
2
configurations,
each
of
energy
1.
Overall,
counting
configurations
of
the
same
energy:
(U=0)
=
3+2+2+3+2+2+3=17
;
(U=1)
=
2+1+1+1+1+2=8
The
number
of
microscopic
configurations
and
energy
levels
increases
rapidly
with
chain
length.
Theoretical
and
Monte
Carlo
computer
simulation
techniques
are
used
for
determining
the
properties
of
models
of
this
type
for
longer
chains.
Figure
3.14
Configurations
for
a
two-dimensional
chain
of
5
beads
A key issue in statistical mechanics is the frequency of occurrence of different
microstates when the overall constraints of a thermodynamic system are specified.
A basic postulate, comparable in importance to the postulate about the existence
of equilibrium states in classical thermodynamics discussed in 1.4 (p. 6), is that
all microstates of a system at a given U, N and V are equally probable. The conse-
quences of this postulate will be fully explored in Chapter 5.
60
Chapter 3. Reversibility and Entropy
The
basic
postulate
of
statistical
mechanics
implies
that
the
probability
of
any
microstate
,
P ,
is
the
same
as
that
of
any
other
microstate
in
the
constant
N ,V ,U
ensemble:
1
P =
at
constant
N,
V
and
U
(3.19)
!
From
this
postulate,
we
can
now
simply
derive
another
famous
expression,
the
Gibbs
entropy
formula,
by
substituting
Eq.
3.19
into
Eq.
3.17:
Gibbs
entropy
formula
S = kB
!
P lnP
(3.20)
all!micro+
states!
The
Gibbs
entropy
formula
can
be
shown
to
be
valid
even
for
systems
not
at
constant
energy
U,
volume
V,
and
number
of
particles
N.
This
is
in
contrast
to
Eq.
3.17,
which
is
only
valid
at
constant
for
microstates
at
constant
U
V
and
N.
For
ex-
ample,
in
5.6
we
prove
Eq.
3.20
for
systems
at
constant
N,
V,
and
T.
Chapter 4. Fundamental Equations
Two key problems in thermodynamics are to relate properties to each other and to
develop ways to measure them accurately in the laboratory. For example, we have
seen in Chapter 2 that the relationship between the heat capacities at constant
volume and at constant pressure for an ideal gas is:
C IG = CVIG + R
! P
It
is
highly
desirable
to
obtain
a
general
relationship
between
the
two
heat
capaci-
ties,
C = (U / T )V and
C P = (H / T )P
,
! V
!
for
non-ideal
gases,
liquids
and
solids.
In
the
present
chapter,
we
develop
a
formal
approach
that
allows
us
to
obtain
such
relationships
between
thermodynamic
de-
rivatives
in
a
systematic
fashion.
An
important
consideration
in
developing
thermodynamic
relationships
and
methods
for
determining
derived
thermodynamic
functions
such
as
the
energy,
enthalpy,
or
entropy,
is
the
preservation
of
information
content
as
one
moves
from
one
function
to
another.
In
the
rest
of
this
chapter,
we
demonstrate
how
all
these
functions,
when
expressed
in
terms
of
their
natural
variables,
are
essential-
ly
equivalent
to
each
other
and
encompass
all
available
information
about
equilib-
rium
states
of
a
system.
These
equivalent,
complete
descriptions
of
thermodynam-
ic
properties
are
called
fundamental
equations
and
are
the
main
topic
of
the
pre-
sent
chapter.
4.1 Thermodynamic Calculus
As already mentioned in Chapter 1, for a given quantity of a one-component sys-
tem at equilibrium, all properties are completely determined by specifying the
values of two additional independent thermodynamic variables. More generally,
n+2 independent thermodynamic variables, where n is the number of components
in a system, are needed to fully characterize equilibrium states for multicompo-
nent systems. Common thermodynamic variables used to specify equilibrium
Draft material from Statistical Thermodynamics, (2012), A.Z. Panagiotopoulos
79
80
Chapter 4. Fundamental Equations
states
in
such
systems
are
the
total
volume
V,
number
of
moles
of
each
component,
N1,
N2,
Nn,
temperature
T
and
pressure
P.
The
total
entropy
S
can
also
be
specified
and
controlled
with
some
effort,
namely
by
performing
reversible
processes
and
using
the
definition
developed
in
Ch.
3,
to
measure
entropy
changes.
S = Q rev /T
!
It is clear from empirical observations that not all combinations of variables
result in a unique specification of the state of thermodynamic systems. This is es-
pecially apparent when the possibility of coexistence between phases of different
properties is taken into account. For example, at a given temperature and at the
saturation pressure in a one-component system, there are infinitely many ways in
which two phases can coexist as equilibrium thermodynamic states differing in the
relative amounts of the two phases. In other words, specifying the pressure and
temperature at vapor-liquid equilibrium of a pure fluid does not uniquely specify
the relative amounts of liquid and vapor. Similarly, a given pressure and volume
(or density) of a binary mixture can be realized in many different ways for a range
of temperatures in a two-phase system. In addition to the issue of the uniqueness
of thermodynamic states, certain privileged sets of variables are natural for spe-
cific properties. Thermodynamic functions expressed in their natural variables
contain more complete information than the same properties expressed in other
sets of variables. In order to explain why this is the case, let us consider the com-
bined statement of the First and Second Laws for closed systems derived in 3.4:
!dU = TdS PdV
(4.1)
This
expression
suggests
that
the
volume
and
entropy
constitute
a
good
set
of
variables
for
describing
the
energy
U
of
a
closed
system.
More
formally,
we
write:
U
U
U = U(S,V ) dU =
dS +
dV
S V
V S
!
(4.2)
The derivatives are identified as:
U
U
S = T
and
V = P
S
!
!
(4.3)
To
obtain
the
full
expression
for
U
as
a
function
also
of
the
amount
of
material
in
a
system
in
addition
to
S
and
V,
we
need
to
augment
Eq.
4.2
by
additional
varia-
bles,
namely
the
amount
of
material
of
each
component
present
in
the
system.
For
this
purpose,
we
define
an
additional
partial
derivative,
written
here
for
a
one-
component
system:
81
4.1 Thermodynamic Calculus
U
(4.4)
N S ,V
!
This
derivative
defines
an
important
new
thermodynamic
variable,
the
chemi-
cal
potential
of
a
component.
The
chemical
potential
is
an
intensive
variable
that
has
units
of
specific
energy,
[J/mol].
Its
physical
interpretation
in
classical
thermo-
dynamics
is
somewhat
obscure
when
compared
to
the
two
other
more
intuitively
understood
first-order
derivatives
of
the
energy
function
given
by
Eqs.
4.3.
How-
ever,
the
chemical
potential
plays
just
as
fundamental
a
role
in
thermodynamics
as
temperature
and
pressure.
An example of a relatively simple instrument that measures chemical poten-
tials is the pH meter, which many of us have used in chemistry laboratories. The
instrument measures the chemical potential of hydrogen ions, not their concentra-
tion. Of course, the two are related, with the chemical potential increasing with
increasing concentration but there is a clear distinction. After all, density also
increases with pressure, but pressure and density are not usually confused to be
the same physical quantity! Another way to develop a physical feeling for the
chemical potential is to consider your senses of smell and taste. What exactly is
being measured when you find that a food tastes sour, or when you smell a fra-
grance from a flower? One may naively suggest that it is the concentration of the
molecules that correlates with the intensity of taste and smell, but in reality it is
the chemical potential of the corresponding components that drives the biochemi-
cal receptors responsible for these senses. In addition to chemical reactions, diffu-
sion and evaporation are also driven by chemical potential (not concentration)
differences. On a hot and humid summer day, sweating does not cool you down
because there are no chemical potential differences to drive evaporation, despite
the large concentration difference for water between wet skin and air.
Using the newly defined derivative of Eq. 4.4, we can now write a complete dif-
ferential relationship for the energy as a function of entropy, volume and number
of moles, U = U (S,V,N):
!dU = TdS PdV + dN
(4.5)
More
generally,
for
a
multicomponent
system,
one
needs
n+2
variables
to
fully
characterize
the
state
of
a
system.
When
the
function
of
interest
is
the
total
energy
U,
these
variables
can
be
selected
to
be
the
total
entropy,
S,
volume,
V
and
amount
of
moles
for
each
component
present,
{N} N1 ,N2 ,Nn :
dU = TdS PdV + i dNi
i=1
!
(4.6)
The chemical potential i of a component in a mixture is defined from a direct
extension of Eq. 4.4,
funda-
mental
equation
82
Chapter 4. Fundamental Equations
U
i
Ni S ,V ,N
ji
!
(4.7)
In
this
equation,
Nji
indicates
that
the
number
of
moles
of
all
components
other
than
i
are
kept
constant;
more
explicitly,
N1 ,N2 , Ni1 ,Ni+1,
Nn
are
constant
while
Ni
varies.
Because
the
chemical
potential
definition
implies
an
open
system
to
which
a
component
is
added,
its
value
depends
on
the
reference
state
for
energy
for
the
corresponding
component
and
thus
is
not
directly
measurable,
unlike
the
first
two
derivatives
of
Eq.
4.6,
temperature
and
pressure.
Eq.
4.6
is
the
single
most
important
equation
of
this
book
and
is
known
as
the
fundamental
equation
of
thermodynamics
(abbreviated
FE).
It
links
the
key
quan-
tities
of
the
First
and
Second
Laws
and
provides
the
starting
point
for
understand-
ing
equilibrium
and
stability
as
well
as
for
deriving
thermodynamic
relations.
S,
V
and
{N},
the
variables
in
which
the
function
U
is
expressed
in
this
representation,
are
all
extensive,
proportional
to
system
size
(mass).
The
first
derivatives,
T,
P
and
i's
are
all
intensive,
independent
of
system
size.
An
equivalent
representation
of
the
entropy
S
as
a
function
S (U ,V ,N1 ,N2 , Nn)
can
be
obtained
by
rearrangement
of
the
terms
of
Eq.
4.6:
n
1
P
dS = dU + dV i dN i
T
T
i=1 T
!
(4.8)
This representation of the fundamental equation is used in statistical mechanics.
Example 4.1
The fundamental equation for a pure substance is
aUV bN 3
S=
N
UV
!
where
a
and
b
are
positive
constants.
Obtain
the
equation
of
state,
P
as
a
function
of
V
and
T,
for
this
material.
Starting
from
the
FE
in
the
entropy
representation
(Eq.
4.8),
we
obtain
the
pres-
sure
and
temperature
as:
1 S
aV bN 3
P S
aU bN 3
=
=
+
a
nd
=
=
+
T U V ,N N U 2V
T V U ,N N UV 2
!
!
(i)
Taking the ratio of the two expressions in (i),
Adapted from Problem 2.3-5 in Callen, H. B, Thermodynamics and Introduction to Thermo-
statistics, 2nd Ed., Wiley (1985).
83
4.1 Thermodynamic Calculus
aU bN 3
+ 2
P /T
= P = N UV 3
1/T
aV bN
+
N U 2V
!
aU 2V 2 + bN 4
U
NUV 2
=
=
U = PV
2 2
4
V
aU V + bN
NU 2V
(ii)
substituting (ii) into (i),
3
4
P aPV bN 3
b
=
+
= aPV + 3 P 2 V aV T = bT
3
T
N
PV
PV
!
bT
P=
V aV T
4.2 Manipulation of Thermodynamic Derivatives
Deriving relationships between thermodynamic derivatives is the key objective of
the present chapter. Manipulations of thermodynamic derivatives can seem quite
daunting at times, but can be accomplished systematically with a small number of
relatively simple mathematical tools. Thermodynamic functions, after all, are
mathematical objects that are subject to the same rules of multivariable calculus as
any other functions, even if we frequently do not have explicit, closed-form ex-
pressions for them in terms of their independent variables.
In the following, we will consider x, y, z, and w to be mathematical objects
with well-behaved functional relationships between them. We will also assume
that there are two independent variables in this set and that any two of x, y, z, and
w can serve as the independent variables. This assumption is made for simplicity
and without loss of generality. The following general mathematical relationships
are often useful for manipulating derivatives of functions of many variables in
general and thermodynamic functions in particular:
Inversion
x
1
=
y / x
y z
!
(4.9)
This
relationship
is
for
the
underlying
functions
rather
than
simple
algebraic
in-
version;
x(y,z)
and
y(x ,z)
that
are
being
differentiated
on
the
two
sides
of
Eq.
!
!
4.9
are
expressed
in
terms
of
different
variables
(see
Example
4.2
for
an
illustra-
tion
of
this
concept).
84
Chapter 4. Fundamental Equations
Commutation
2z
z
z
=
(4.10)
=
x y
xy
x
y x y
x
y
!
In
other
words,
mixed
second
derivatives
are
independent
of
the
order
in
which
they
are
taken.
This
property
applied
to
thermodynamic
functions
leads
to
equali-
ties
between
derivatives
known
as
Maxwells
relations.
Chain
rule
(
(
)
)
x / w
x
x w
z
=
=
(4.11)
w y
y
y
/
w
z
z !!!inversion!
z
z
!!!!!!!!rule!!!
!
In
Eq.
4.11
we
have
changed
the
independent
variables
from
x(y,z)
in
the
left-
!
hand-side
to
x(w,z)
and
y(w,z)
in
the
right-hand-side.
!
!
Triple-product rule (also known as XYZ-1 rule)
This relationship links three cyclically permuted derivatives:
x y z
= 1
y z z x x y
!
(4.12)
Here,
the
three
functions
being
differentiated,
x(y,z) ,
y(x ,z) ,
and
z(x ,y) ex-
!
!
!
press
the
same
underlying
relationship
in
terms
of
different
variables.
To
prove
this
equality,
let
us
consider
the
differential
form
of
the
function
x = x(y,z) :
x
x
dx =
dy +
dz
z y
y z
!
at constant x, this expression becomes:
x
x
dy x +
dz x = 0
z y
y z
!
Now let us obtain the derivative / z
!
of
this
relationship:
x y
x y
x
x
=0
+
=
z y
y z z x z y
y z z x
!
85
4.2 Manipulation of Thermodynamic Derivatives
x y z
= 1
y z z x x y
!
Example
4.2
Demonstrate
how
the
inversion,
commutation,
chain
and
triple-product
rules
ap-
ply
to
the
functions:
y2
x(y,z) =
;
w = xz
(variable
transformation
for
the
chain
rule)
z
!
For
the
function
x
in
the
original
representation,
x = x(y,z) :
!
x / y = 2y /z
z
!
To
test
the
inversion
rule,
we
obtain
the
function
in
the
representation
y = y(x ,z) :
!
y
y2
z
x=
y 2 = xz y = xz
=
z
x z
2 x
!
Now we need to eliminate x :
z
2 x
z
2 y 2 /z
z
1
=
2y x / y
y
1
=
(x / y)z
x z
!
For the commutation rule (independence on the order of differentiation), we have
2
2x 2y
2y
y
2 = 2
= =
z
yz z z y y z z
!
For
the
chain
rule,
the
new
variable
is
defined
as
w = xz .
Then:
w
1
x / w =
z
z
z
2
y
w = xz =
z y = w y / w
z
!
x=
So that
( x / w )
( y / w )
!
1/z
1/2 w
2y
z
= x / y
1
2 w
86
Chapter 4. Fundamental Equations
For the triple-product rule we obtain:
x
x=
= 2y /z
z
y z
y2
y
y
x
y = xz
=
=
z x 2 z 2z
z=
!
y2
z
y2
z2
x
x y
x2
y2
x y z
2y y z 2
!
!
=
= 1
2
y
z
x
z
2z
y
z
x
One
could
have
substituted
variables
other
than
y
and
z
in
the
expressions
for
the
three
cyclic
derivatives
with
identical
results.
4.3 Eulers Theorem for Homogeneous Functions
Thermodynamic functions and derivatives have special properties resulting from
the fact that they are either extensive (proportional to the size of the system) or
intensive (independent of the size of the system). Mathematically, functions that
satisfy the relationship
f (x) = f (x) for any x
!
are called homogeneous of degree one. Functions can be homogeneous of degree
one with respect to some, but not all the variables for example, a function that is
homogeneous of degree one with respect to variables x1,x2, ,xi and homogenous
of degree zero with respect to variables y1,y2, ,yj has the property:
f (x1 ,xi ,y1 ,y j ) = f (x1 ,xi ,y1 ,y j )!
!!for!any!!x1 ,xi ,y1 ,y j
(4.13)
!
Extensive
thermodynamic
functions
are
homogeneous
of
degree
one
with
respect
to
their
extensive
variables,
and
homogeneous
of
degree
zero
with
respect
to
their
intensive
variables.
Eulers
theorem
for
homogeneous
functions
provides
a
link
between
such
func-
tions
and
their
derivatives,
as
follows:
!
f
f
f
f (x1 ,xi ,y1 ,y j ) = x1
+ x2
++ xi
x1
x2
xi
The proof of this theorem is as follows.
(4.14)
87
4.3 Eulers Theorem for Homogeneous Functions
f (x1 ,xi ,y1 ,)
!
=
f (x1 ,xi ,y1 ,)
f (x1 ,xi ,y1 ,) (x1 )
(x1 )
f (x1 ,xi ,y1 ,)
++
= f (x1 ,xi ,y1 ,)
f (x1 ,xi ,y1 ,) (xi )
(xi )
f (x1 ,xi ,y1 ,)
=
x1 + +
xi
x1
xi
!
Application
of
Eulers
theorem
to
U (S ,V ,N1,N2, ,Nn),
a
homogeneous
function
of
degree
one
with
respect
to
all
its
variables,
gives:
U
U
U = S
+ V
+
S V ,{N }
V S ,{N }
!
Ni N
i=1
S ,V ,N
ji
U = TS PV + i Ni
i=1
!
(4.15)
This Euler-integrated form of the fundamental equation is sometimes confusing, as
it superficially appears to suggest that U is a function of twice as many variables as
previously (T and S, P and V, i and Ni). This is incorrect; despite appearances to
the contrary, the same variable set as before, ( S ,V ,N1,N2, ,Nn), is being used, with
temperature, pressure and chemical potentials that appear in the equation also
being functions of the same variables: T (S ,V ,N1,N2, ,Nn), P (S ,V ,N1,N2, ,Nn), and
i (S ,V ,N1,N2, ,Nn).
4.4 Legendre Transformations
Even though it is possible in principle to control the variables ( S ,V ,N ) experimen-
tally, it is much more common to perform experiments under constant-tempera-
ture or constant-pressure conditions, rather than at constant volume or entropy.
For example, constant-temperature conditions are closely approximated when a
system is immersed in a temperature-controlled bath. Constant-pressure condi-
tions are imposed when a system is open to the atmosphere. Variables such as T
and P appear as the first derivatives of the fundamental equation U (S ,V ,N ). Unfor-
tunately, while it is certainly possible to obtain functions such as U (T ,P ,N ), the
information content of such functions is not equivalent to that of the fundamental
equation U (S ,V ,N ) this is because of the integration constants that will need to
be introduced if one wants to go from U (T ,P ,N ) to U (S ,V ,N ), as illustrated in Ex-
ample 4.3.
Euler-
integrated
FE
88
Chapter 4. Fundamental Equations
In Chapter 2, we have already introduced an additional thermodynamic func-
tion, the enthalpy H = U + PV. Here, we will show that this and other similar de-
rived thermodynamic functions are more than just convenient combinations of
terms. They represent fundamental equations that play the same key role in ther-
modynamics as the energy U, but are expressed in variables other than ( S ,V ,N ).
In mathematical terms, for a function f(x ) of a single variable x, we would
like to obtain a new function g ( ), with the same information content as the origi-
nal function, but expressed in a new variable = df/ dx, the first derivative of the
original function. Equivalence of information content implies that the original
function can be recovered from the transformed one without any ambiguity in the
form of integration constants. It turns out that this can be done through a mathe-
matical operation known as a Legendre transformation. For a function of one vari-
able, the transformed function is obtained as g ( ) = f x , which has the following
properties:
d f = dx
dg = df xd dx = xd
!
(4.16)
In
the
new
function
(referred
to
as
the
transform
of
the
original
function),
the
role
of
variables
and
derivatives
has
been
exchanged:
the
derivative
of
the
original
function
is
the
variable
of
the
new
function
and
the
variable
of
the
original
function
is
minus
the
derivative
of
the
transform.
One
can
recover
the
original
function
by
simply
applying
the
Legendre
transformation
one
more
time:
dg = xd
f = g + x
(4.17)
Example
4.3
Consider
the
function:
f (x) = x 2 + 2
Construct
the
function
f(w )
and
show
that
it
cannot
be
used
to
fully
determine
f(x ).
Also
obtain
the
Legendre
transform
of
this
function,
g (w )
and
show
how
it
can
be
used
to
reconstruct
the
original
function
f (x ).
The
derivative
of
f (x ) is:
!
df
=
= 2x
dx
!
The
function f( )
can
be
obtained
by
substituting
x
=
/ 2
in
f (x ):
89
4.4 Legendre Transformations
f () =
2
+ 2
4
(i)
Given f( ), we can attempt to determine f(x ) from:
2
df
dx 1
1
=
x = df =
= f 2 + c f = x c + 2
df
2 f 2
(ii)
An unknown integration constant c appears in expression (ii), so we have shown
that f(x ) cannot be fully recovered from f( ).
The Legendre transform g ( ) of f(x ) is:
2
g() = f x = + 2 g() = + 2
4
2
4
!
(iii)
From Eq. 4.17, the variable x can be obtained from (iii) from differentiation:
dg
x=
=
d 2
!
Now
the
function f(x )
can
be
obtained
from
g ( )
using
Eq.
4.17
again:
f = g + x =
(iv)
2
+ 2+ w = + 2 f = x 2 + 2
QED
4
2
4
Example
4.4
Consider
the
function
f
(
x
)
=
x 3
+
1
in
the
interval
[1,1].
Construct
its
first
Legen-
dre
transformation g ( )
and
plot f (x
)
and g ( ),
marking
the
points
correspond-
ing
to
x
=
0.7, x
=
0,
and
x
=
0.7
on
the
graph
of g ( ).
Using
the
definition
of
Legendre
transforms:
df
= = 3x 2 x =
3
! dx
3/2
23/2
3/2
3/2
+1
g() = f x = +1
=
+1
g() =
3 3 3
3
3
3
3
!
!
The
Legendre
transform
of
g ( )
is:
dg
=
= x
3
! d
90
Chapter 4. Fundamental Equations
dg
23/2
3/2
=
+1
=
+1 = +1 = x 3 +1 = f (x)
g
d
3 3 3
3 3
3
!
Plots
of
the
original
and
transformed
functions
are
shown
in
Fig.
4.1.
Arrows
mark
the
three
points
of
interest
(note
that
=
3x 2,
so
the
coordinate
corresponding
to
x
=
0.7
is
=
30 .7 2
=
1.47.
The
transform
is
multivalued
two
values
of
the
function
g ()
exist
for
any
given
.
This
is
the
price
we
have
to
pay
for
ensuring
that
the
reverse
transform
is
unique.
In
addition,
the
transformed
function
has
a
cusp
at
=0,
with
a
discon-
tinuous
first
derivative.
Multivalued
functions
arise
naturally
in
thermodynamics
when
a
system
can
exist
in
multiple
phases;
cusps
are
also
present
in
thermody-
namic
functions
for
systems
with
multiple
phases.
Figure 4 .1 A one-dimensional function and its transform.
For
functions
of
many
variables,
the
Legendre
transform
procedure
can
be
applied
sequentially,
giving
the
first,
second
and
higher-order
transforms.
Formally,
we
consider
a
general
function
of
l
variables,
as
a
basis
function
y (0) ,
where
basis
!
means
simply
the
starting
point
of
the
transform:
y (0)(x1 ,x2 ,,xk ,xk+1 ,xl )
!
The first and second derivatives of the basis function are defined as:
(4.18)
91
4.4 Legendre Transformations
i y (0)
i
y (0)
=
x i
!!!!and!!!!!!!!y (0)
ij
x j[i ]
(0)
2y (0)
y
!
=
=
(4.19)
x i x j
xi x j
xi[ j ]
x
j[i ]
Thus, the differential form of y (0) is:
!
dy (0) = 1x1 + 2x2 + + kxk + k+1xk+1 + lxl
(4.20)
!
The k-th transform of y (0) is denoted as y (k ) and is obtained from:
!
!
(k)
y (1 ,2 ,, k ,xk+1 ,xl ) = y (0) x11 x22 xk k (4.21)
!
The k-th transform is a function of a new set of variables, (1 ,2 ,, k ,xk+1 ,xl ) .
!
Its differential form is
dy (k) = x1d1 x2d2 xk d k + k+1dxk+1 + + l dxl (4.22)
!
A common basis function for thermodynamics is the fundamental equation in
the energy representation, U (S ,V ,N1,N2, ,Nn). Additional thermodynamic func-
tions, such as H, are simply transforms of the fundamental equation U that pre-
serve its information content. Note that different Legendre transforms result if the
order of variables of the original function is changed, for instance to
U (V ,S ,N1,N2, ,Nn).
Fundamental!equation:!!y (0) = U(S,V ,N1 ,N2 ,,Nn )
dU = TdS PdV + i dNi
(4.5)
i=1
The
first
Legendre
transformation
of
U(S,V ,N1 ,N2 ,,Nn )
gives
a
new
thermo-
!
dynamic
function,
the
Helmholtz
free
energy
A:
Fundamental!equation:!!y (1) = A(T ,V ,N1 ,N2 ,,Nn )
A = U TS ; dA = SdT PdV + i dNi
(4.23)
i=1
The
second
transform
of
U,
gives
another
fundamental
equation,
the
Gibbs
free
energy
G,
named
after
American
scientist
Josiah
Willard
Gibbs
(1839-1903):
Fundamental!equation:!!y (2) = G(T ,P,N1 ,N2 ,,Nn )
G = U TS + PV = A + PV ; dG = SdT +VdP + i dNi
i=1
!
(4.24)
92
Function
S
V
N
Internal energy U
!dA = SdT PdV + dN
V N
Helmholtz energy A
!dH = TdS +VdP + dN
S P N
Enthalpy H
!dG = SdT +VdP + dN
P N
Gibbs free energy G
Table 4.1 Fundamental equations (thermodynamic potential functions) for one-component systems.
Variables
!dU = TdS PdV + dN
!G = N
Differential
!H = TS + N
G
G
S=
V =
T P ,N
P T ,N
!
!
G
=
= G
N T ,P
!
H
H
T=
V =
S P ,N
P S ,N
!
!
H
=
N S ,P
V
T
=
S P ,N P S ,N
!
S
V
=
P T ,N T P ,N
2
2
NC P
S
T
T
H
G
=
=
= =
S 2 P ,N S P ,N NC P
T 2 P ,N T P ,N T
!
!
!A = PV + N
P
S
=
T V ,N
V T ,N
!
2
NCV
S
A
= =
T 2 V ,N T V ,N T
!
U
U
A
A
S =
T=
P=
P =
S V ,N
V S ,N
T V ,N
V T ,N
!
!
!
!
U
A
=
=
N S ,V
N T ,V
!
P
T
=
S V ,N V S ,N
2
T
T
U
= =
S 2 V ,N S V ,N NCV
!
!U = TS PV + N
Integral
First
de-
rivatives
A
second
derivative
Maxwells
rule
93
4.4 Legendre Transformations
Reordering
the
variables
to
U(S,V ,N1 ,N2 ,,Nn )
gives
the
enthalpy
H,
first
in-
!
troduced
in
deriving
First
Law
balances
for
open
systems
as
the
first
Legendre
(1)
transform
y :
!
Fundamental!equation:!!y (1) = H(S ,P,N1 ,N2 ,,Nn )
H = U + PV ; dH = TdS +VdP + i dNi
(4.25)
i=1
The Legendre transforms of U are also known as thermodynamic potentials.
Table 4.1 (p. 92) summarizes the information on variables and derivatives for the
four thermodynamic potentials we have discussed thus far, for the case of a one-
component system for simplicity. The table also lists a second derivative for each
transform and an example Maxwells relationship between second derivatives.
The Euler-integrated form of each of the fundamental equations expresses
them as sum of terms, each term being an extensive variable of the transform mul-
tiplied by the corresponding derivative. The results are summarized in Table 4.1
and are consistent with the Legendre transformations above. For example, for the
Gibbs free energy G,
!G = N = U TS + PV = H TS = A + PV
(4.26)
An important consequence of the integral relationship for G in the special case of a
one-component system is that the molar Gibbs free energy is, in this case, equal to
the chemical potential:
for one-component systems only: !G =
(4.27)
Additional
transformations,
not
listed
in
the
table,
are
possible
as
well.
For
ex-
ample,
one
could
order
the
variables
as
U(N1 ,N2 ,,Nn ,S,V )
and
perform
a
single
!
transform
to
obtain
a
function
of
(1 ,N2 ,,Nn ,S,V ) .
The
resulting
function
is
also
!
a
valid
fundamental
equation,
even
if
it
is
not
frequently
used
in
practice.
The
final
comment
here
is
that
the
last
transform,
y (n+2) ,
yields
a
function
of
!
only
intensive
variables,
T,
P
and
all
the
chemical
potentials.
By
Euler
integration
we
obtain
that
the
resulting
function
is
identically
zero,
a
result
known
as
the
Gibbs-Duhem
relationship:
n
SdT +VdP Ni d i = 0
i=1
!
(4.28)
Gibbs-
Duhem
relationship
4.5 Derivative Relationships
As already stated, two key objectives of classical thermodynamics are to obtain
relationships between properties, and to develop techniques to measure them. Ob-
taining thermodynamic relationships helps bring forward hidden connections
between seemingly unrelated quantities and facilitates the measurement and vali-
dation of physical property data.
The Legendre transformation facilitates developing relationships between
thermodynamic derivatives. For example, the following relationships exist be-
tween second derivatives of a transform and its basis function:
(1)
y11
=
!
y1i(1) =
y (1)
ij
!
y (0)
ij
y1i(0)
(0)
y11
1
(0)
y11
!!!!!i 1
(0) (0)
y1i
y1j
(0)
y11
!!!!!i, j 1
(4.29)
(4.30)
(4.31)
For proofs of these relationships, as well as more complex general relation-
ships between derivatives of the k-th transform and those of the basis function, see
Beegle, Modell and Reid, AIChE J., 20: 1194-200 (1974) and Kumar and Reid, AIChE
J., 32: 1224-6 (1986).
For pure components, a major simplification of thermodynamic relationships
is possible relative to the general multicomponent case, because composition is
not an independent variable. Extensive properties can then be normalized by the
amount of mass (moles) in a system to obtain intensive properties. Thermodynam-
ic derivatives at constant number of moles can be taken either on a total or on a
molar basis, for example:
U
(NU)
U
=
=
S
= T
V ,N (N S) NV ,N S V
!
(4.32)
N
was
eliminated
in
the
last
derivative
of
this
expression
because
intensive
prop-
erties
do
not
depend
on
the
size
of
the
system,
so
that
N
is
no
longer
a
relevant
constraint.
The
differential
forms
of
the
fundamental
equations
in
one-component
systems
for
the
intensive
thermodynamic
functions
U,
A,
G,
and
H,
are
as
follows:
94
4.5 Derivatives in Terms of Measurable Properties
95
dU = TdS PdV
d A = SdT PdV
dH = TdS +VdP
!dG = SdT +VdP
(4.33)
First
derivatives
of
these
functions
with
respect
to
their
natural
variables
are
obtained
directly,
either
from
Table
4.1
or
from
Eqs.
4.33.
An
example
of
such
a
first
derivative
is
given
above
in
Eq.
4.32
temperature
is
clearly
an
experimental-
ly
measurable
quantity.
Other
examples
include:
U
U
H
G
=
= P
and
= = V
V
S ,N V S
P S P T
!
!
(4.34)
Some first derivatives of Fundamental Equations with respect to natural varia-
bles cannot be measured directly:
U
A
G
T = T = S
and
N =
S ,V
V P
!
!
(4.35)
First derivatives of Fundamental Equations with respect to unnatural varia-
bles or under other constraints can be obtained from the differential expressions
of Eq. 4.33. For example, to obtain the volume derivative of the energy at constant
temperature in terms of measurable properties, we start from:
!dU = TdS PdV
We now take the partial derivative ( / V )T , followed by Maxwells rule on A to
!
obtain:
U
S
P
V = T V P = T T P
T
T
V
!
(4.36)
Second derivatives of fundamental equations with respect to their natural var-
iables are important from a theoretical and experimental viewpoint. For example,
differentiating A and G twice with respect to temperature, we obtain two im-
portant experimentally measurable quantities, the heat capacities defined in Chap-
ter 3:
2 A
2 =
T V
!
C
S
1 U
= = V
T T V
T
T V
(4.37)
intensive
FEs
for
one-
component
systems
96
Chapter 4. Fundamental Equations
2 G
C
S
1 H
= P
2 = =
T T P
T
T P
T P
!
(4.38)
The
other
two
second
derivatives
of
G
with
respect
to
its
natural
variables
are
also
important
experimentally
measurable
quantities.
The
cross
derivative
with
respect
to
temperature
and
pressure
is:
!
V
2 G
G
=
=
= PV
TP
T P T
T P
P
(4.39)
This derivative is proportional to the coefficient of thermal expansion, defined as:
(V / T )P /V
! P
(4.40)
The second derivative with respect to pressure is:
2 G
V
2 =
= T V
P T P T
!
(4.41)
This derivative is proportional to the isothermal compressibility, which is defined
as:
(V / P)T /V
! T
(4.42)
Mixed second derivatives of fundamental equations with respect to their natu-
ral variables can be written in two equivalent ways, using the commutation prop-
erty (Maxwells relationships):
P
T
2U
= =
SV
S V
V S
(4.43)
P
S
2 A
=
=
T V
T V
V T
(4.44)
T
V
2H
=
=
S P
P S
S P
(4.45)
S
V
2G
= =
T P
P T
T P
(4.46)
!
It
is
clear
from
the
last
three
expressions
that
derivatives
of
the
entropy
S
with
re-
spect
to
volume
or
pressure
are
experimentally
measurable,
even
though
the
abso-
lute
value
of
entropy
itself
is
not
directly
obtainable
in
classical
thermodynamics.
4.5 Derivatives in Terms of Measurable Properties
97
Other
derivatives
can
be
obtained
using
the
tools
described
in
4.2.
For
exam-
ple,
the
Joule-Thompson
coefficient,
(T/P) H
is
the
rate
of
temperature
change
with
pressure
when
a
fluid
flows
across
a
throttling
valve,
as
can
be
confirmed
from
a
differential
First
Law
balance
in
an
open
system
at
steady
state
with
d/Q
=
d/W
=
0
(see
Eq.
2.18,
p.
18).
This
coefficient
can
be
expressed
in
terms
of
equation-
of-state
derivatives
and
heat
capacities
by
first
invoking
the
triple-product
rule:
(H / P)T
T H P
T
P T H = 1 P = (H / T )
H
P
T
H
P
!
The
denominator
is
simply
CP
;
the
numerator
can
be
obtained
from
the
differential
form
of
H,
H
S
V
dH = TdS +VdP
= T +V = T +V
P T
P T
T P
!
The final result is:
T
1 V
=
T
V
P
C P T P
H
(4.47)
Example 4.5 Differential expressions for U
Obtain differential expressions for U = U ( T ,V ) and U = U ( T ,P ) in terms of experi-
mentally measurable properties. Note that these expressions are not fundamental
equations because the natural variables of U are S and V.
We seek to construct expressions of the form:
U
U
dU = dT + dV
T V
V T
!
(i)
U
U
dU =
dT +
dP
T P
P T
!
(ii)
The temperature derivative (U /T) V = CV , and the volume derivative (U/V) T ,
have been obtained in Eq. 4.36, so the desired expression for U = U (T,V) is:
dU = CV dT + T P dV
T
V
!
(iii)
98
Chapter 4. Fundamental Equations
For
the
derivatives
needed
for
expression
(ii),
we
start
again
from
the
fundamental
equation
for
U
in
terms
of
its
natural
variables,
dU
=
TdS
PdV
.
Differentiating
with
respect
to
T
at
constant
P
gives:
U
S
V
(iv)
T = T T P T
P
P
P
!
The
temperature
dependence
of
the
entropy
at
constant
pressure,
(S
/ T) P
,
can
be
obtained
by
differentiating
dH
=
TdS
+
VdP:
H
S
T = C P = T T
P
P
!
After
substituting
the
result
into
(iv),
we
get:
U
V
T = C P P T
P
P
!
The
pressure
derivative
needed
for
(ii)
is:
U
S
V
V
V
P = T P P P = T T P P
T
T
T
P
T
!
(v)
(vi)
We
used
Maxwells
relationship
on
G
to
get
rid
of
S.
Substituting
(v)
and
(vi)
into
(ii)
we
finally
obtain:
V
V
V
dU = C P P
+ P
dT T
dP
T P
P T
T P
!
(vii)
One
observation
we
can
make
here
is
that
the
functional
forms
of
U
in
unnatural
variables
are
more
complex
than
the
comparable
expressions
in
its
natural
vari-
ables.
This
is
an
additional
disadvantage
beyond
the
loss
of
information
content
discouraging
the
use
of
functional
forms
such
as
!U = U(T ,V )
and
!U = U(T ,P) .
Example
4.6
Difference
between
heat
capacities
Calculate
the
difference
between
the
heat
capacities
at
constant
pressure
and
con-
stant
volume,
C P C V ,
for
a
general
thermodynamic
system,
in
terms
of
P,
V,
T
and
their
mutual
derivatives.
We
use
as
starting
point
expression
(iii)
in
Example
4.5:
dU = CV dT + T P dV
T
V
!
4.5 Derivatives in Terms of Measurable Properties
99
Differentiating
with
respect
to
T
at
constant
P
and
setting
the
result
equal
to
ex-
pression
(v)
of
Example
4.5,
we
obtain:
U
=
C
T
P !!!!!!from!(iii)!!! V
!
P
V
V
+ T P
=
C P P
T
T !!!from!(v)!!!
T P
V
P
P V
C P CV = T
T V T P
!
For
ideal
gases,
one
can
simplify
this
relationship
considerably:
(P / T )V = R /V !!!!and!!!!(V / T )P = R / P C P CV = TR2 /(PV ) = R
!
Thus, the difference in heat capacities at constant pressure and constant volume
for ideal gases is simply the ideal-gas constant R, as already derived in 2.4.
Chapter 5. Statistical Mechanical Ensembles
Boltzmanns microscopic expression for the entropy was introduced in 3.6:
S = kB ln(N ,V ,U)
!
(3.17)
where is the number of distinct microscopic states (microstates). The concept of
microstates arises naturally in quantum mechanics, but can be also introduced in
classical systems, if positions and velocities can be grouped so that, when their
values differ less than some selected (but arbitrary) small value, then they are con-
sidered to be equal. This quantization effect also arises naturally in computations,
which are performed with a finite numerical accuracy, so that two quantities can-
not differ by less than the machine precision.
Boltzmanns entropy formula links a macroscopic thermodynamic quantity,
the entropy, to microscopic attributes of a system at equilibrium. In this section,
we introduce many similar relationships for systems under constraints different
than (N,V,U), in a manner quite analogous to the introduction of fundamental equa-
tions in different variables developed in the previous chapter through the Legen-
dre transform formalism.
The branch of physical science that aims to connect macroscopic properties
and microscopic information about a system is called Statistical Mechanics. The
central question in Statistical Mechanics can be phrased as follows: If particles (at-
oms, molecules, electrons, nuclei, or even living cells) obey certain microscopic
laws with specified interparticle interactions, what are the observable properties
of a macroscopic system containing a large number of such particles? Unlike classi-
cal thermodynamics, statistical mechanics does require input information on the
microscopic constitution of mater of interest, as well as the interactions active
among the microscopic building blocks. The advantage of the approach is that
quantitative predictions of macroscopic properties can then be obtained, rather
than simply relationships linking different properties to each other. Another im-
portant difference between statistical mechanics and macroscopic thermodynam-
ics is that fluctuations, which are absent by definition in the thermodynamics, can
be quantified and analyzed though the tools of statistical mechanics. Fluctuations
are temporary deviations of quantities such as the pressure or energy of a system
from their mean values, and are important in small systems e.g. those studied by
Draft material from Statistical Thermodynamics 2012, A. Z. Panagiotopoulos
9/17/12
version
121
122
Chapter 5. Statistical Mechanical Ensembles
computer simulations, or present in modern nanoscale electronic devices and bio-
logical organelles.
5.1 Phase Space and Statistical Mechanical Ensembles
Postulate I states that macroscopic systems at equilibrium can be fully characterized
by n+2 independent thermodynamic variables. For a 1-component isolated system,
these variables can always be selected to be the total mass N, total volume V and
total energy U. However, at the microscopic level, molecules are in constant motion.
Adopting temporarily a classical (rather than quantum mechanical) point of view,
we can describe this motion through the instantaneous positions and velocities of
the molecules. Examples of microscopic and macroscopic variables are given below
for N molecules of a one-component monoatomic gas obeying classical mechanics.
Microscopic variables (in 3 dimensions)
Macroscopic variables
3N position coordinates (x, y, z)
3N velocity components (ux , uy , uz )
3 independent thermodynamic varia-
bles, e.g., N, V, and U.
Given
that
N
is
of
the
order
of
Avogadros
number
[NA
=
6.02211023
mol1]
for
macroscopic
samples,
there
is
a
huge
reduction
in
the
number
of
variables
re-
quired
for
a
full
description
of
a
system
when
moving
from
the
microscopic
to
the
macroscopic
variables.
Moreover,
the
microscopic
variables
are
constantly
chang-
ing
with
time,
whereas
for
a
system
at
equilibrium,
all
macroscopic
thermodynam-
ic
quantities
are
constant.
The
multidimensional
space
defined
by
the
microscopic
variables
of
a
system
is
called
the
phase
space.
This
is
somewhat
confusing,
given
that
the
word
phase
has
a
different
meaning
in
classical
thermodynamics
the
term
was
introduced
by
J.
Willard
Gibbs,
no
stranger
to
the
concept
of
macroscopic
phases.
In
general,
for
a
system
with
N
molecules
in
3
dimensions,
phase
space
has
6N
independent
variables
(r N ,pN ) (r1 ,r2 ,,rN ,p1 ,p2,pN )
(5.1)
!
where bold symbols indicate vectors, ri is the position and pi the momentum of the i-
the molecule, (pi = mi ui). The evolution of such a system in time is described by
Newton's equations of motion:
9/17/12 version
123
5.1 Phase Space and Statistical Mechanical Ensembles
dri
= ui
dt
du i
U(r1 ,r2 ,,rN )
mi
=
dt
ri
!
(5.2)
where U
is
the
total
potential
energy,
which
includes
interactions
among
molecules
and
any
external
fields
acting
on
the
system.
For
a
much
simpler
system,
a
one-dimensional
harmonic
oscillator
shown
in
Fig.
5.1,
phase
space
is
two-dimensional,
with
coordinates
the
position
and
the
momentum
variables.
In
the
absence
of
friction,
the
oscillator
undergoes
harmonic
motion,
which
can
be
represented
as
a
circle
in
phase
space
if
position
and
momentum
are
scaled
appropriately.
The
diameter
of
the
circle
depends
on
the
total
energy
(a
constant
of
the
motion),
and
the
oscillator
moves
around
the
circle
at
a
constant
velocity.
Figure
5.1
A
one-dimensional
har-
monic
oscillator
(top),
and
the
cor-
responding
phase
space
(bottom).
A statistical mechanical ensemble is a collection
of all microstates of a system, consistent with
the constraints with which we characterize a system macroscopically. For example,
a collection of all possible states of N molecules of gas in the container of volume V
with a given total energy U is a statistical mechanical ensemble. For the frictionless
one-dimensional harmonic oscillator, the ensemble of states of constant energy is
the circular trajectory in position and momentum space shown in Fig. 5.1 bottom.
5.2 Molecular Chaos and Ergodic Hypothesis
What causes the huge reduction from 3N time-
dependent coordinates needed to fully character-
ize a system at the molecular level to just a hand-
ful of time-independent thermodynamic variables
at the macroscopic level? The answer turns out to
be related to the chaotic behavior of systems with
many coupled degrees of freedom.
To illustrate this concept, one can perform a
thought experiment on the system shown in Fig.
5.2. In this system, a number of molecules of a gas
are given identical velocities along the horizontal
coordinate, and are placed in an insulated box
9/17/12 version
Figure
5.2
A
conceptual
exper-
iment
in
an
isolated
system.
124
Chapter 5. Statistical Mechanical Ensembles
with
perfectly
reflecting
walls.
Such
a
system
would
seem
to
violate
the
classical
thermodynamics
postulate
of
eventually
approaching
an
equilibrium
state.
Since
there
is
no
initial
momentum
in
the
vertical
direction,
Newtons
equations
of
mo-
tion
would
suggest
that
the
molecules
will
never
hit
the
top
wall,
which
will
thus
experience
zero
pressure
at
all
times,
even
though
there
is
a
finite
density
of
gas
in
the
system.
In
statistical
mechanical
terms,
microstates
with
non-zero
vertical
momenta
will
never
be
populated
in
such
a
system.
Of
course,
even
tiny
interac-
tions
between
the
molecules,
or
minor
imperfections
of
the
walls,
will
eventually
result
in
chaotic
motion
in
all
directions;
thermodynamic
equilibrium
will
be
es-
tablished
over
times
significantly
greater
than
the
interval
between
molecular
col-
lisions.
Most
molecular
systems
are
able
to
forget
their
initial
conditions
and
evolve
towards
equilibrium
states,
by
sampling
all
available
phase
space.
Only
non-
equilibrium
(for
example,
glassy)
systems
violate
this
condition
and
have
proper-
ties
that
depend
on
their
history
such
systems
cannot
be
analyzed
with
the
methods
of
equilibrium
thermodynamics
or
statistical
mechanics.
At
a
molecular
level,
the
statement
equivalent
to
Postulate
I
of
classical
ther-
modynamics
is
the
ergodic
hypothesis.
The
statement
is
as
follows.
Ergodic
hypothesis
For
sufficiently
long
times,
systems
evolve
through
all
microscopic
states
con-
sistent
with
the
external
and
internal
constraints
imposed
on
them.
Experimental
measurements
on
any
macroscopic
system
are
performed
by
ob-
serving
it
for
a
finite
period
of
time,
during
which
the
system
samples
a
very
large
number
of
possible
microstates.
The
ergodic
hypothesis
suggests
that
for
long
enough
times,
the
entire
ensemble
of
microstates
consistent
with
the
microscopic
constraints
on
the
system
will
be
sampled.
A
schematic
illustration
of
trajectories
of
ergodic
and
non-ergodic
systems
in
phase
space
is
shown
in
Fig.
5.3
a
two-
dimensional
representation
of
phase
space
is
given,
whereas
we
know
that
for
re-
alistic
systems
phase
space
has
a
very
large
number
of
dimensions.
The
interior
of
the
shaded
region
is
the
phase
space
of
the
corresponding
system.
For
the
system
Figure
5.3
Schematic
trajectories
of
ergodic
(left)
a nd
non-ergodic
(right)
systems
in
phase
space.
9/17/12 version
125
5.2 Molecular Chaos and Ergodic Hypothesis
on
the
left,
there
are
two
connected
regions
of
phase
space,
so
the
trajectory
even-
tually
passes
from
one
to
the
other
and
samples
the
whole
of
phase
space.
By
con-
trast,
for
the
system
on
the
right,
the
two
regions
of
phase
space
are
disconnected,
so
that
the
system
cannot
pass
from
one
to
the
other.
The
system
is
non-ergodic.
As
already
discussed
in
Chapter
1,
there
is
a
link
between
time
scales
and
con-
straints;
a
system
observed
over
a
short
time
can
appear
to
be
isolated,
while
over
longer
times
it
may
exchange
energy
and
mass
with
its
surroundings.
Also,
the
possible
microstates
depend
on
the
time
scales
of
interest,
and
on
internal
con-
straints
for
example,
chemical
reactions
open
up
additional
microstates,
but
can
only
occur
over
long
time
scales,
or
in
the
presence
of
a
catalyst.
For
systems
satisfying
the
ergodic
hypothesis,
experimental
measurements
(performed
by
time
averages)
and
statistical
mechanical
ensemble
averages
are
equivalent.
Of
course,
we
have
not
specified
anything
up
to
this
point
about
the
rel-
ative
probabilities
of
specific
states;
the
ergodic
hypothesis
just
states
that
all
states
will
eventually
be
observed.
A
general
property
F
can
be
formally
written
as:
Fobserved
!
P
!!!!!probability!of!finding
the!system!in!microstate!
= < F >
value!of!property!F
!!!!!in!microstate!
ensemble
!!average
(5.3)
The
objective
of
the
next
few
sections
will
be
to
determine
the
relative
probabilities,
P ,
of
finding
systems
in
given
microstates
of
ensembles
under
vary-
ing
constraints.
This
will
allow
the
prediction
of
properties
by
performing
ensem-
ble
averages,
denoted
by
the
angle
brackets
of
the
rightmost
side
of
Eq.
5.3.
5.3 Microcanonical Ensemble: Constant U, V, and N
The simplest set of macroscopic constraints that can be imposed on a system are
those corresponding to isolation in a rigid, insulated container of fixed volume V.
No energy can be exchanged through the boundaries of such a system, so Newtons
equations of motion ensure that the total energy U of the system is constant. For
historical reasons, conditions of constant energy, volume, and number of particles
(U, V, N) are defined as the microcanonical ensemble. In the present section (and
the one that follows), we will treat one-component systems for simplicity; general-
ization of the relationships to multicomponent systems is straightforward.
How can we obtain the probabilities of microstates in a system under constant
U, V, and N? Consider for a moment two microstates with the same total energy,
depicted schematically in Fig. 5.4. One may be tempted to say that the microstate
on the left, with all molecules having the same velocity and being at the same hori-
zontal position is a lot less random than the microstate on the right, and thus less
likely to occur. This is, however, a misconception akin to saying that the number
9/17/12 version
126
Chapter 5. Statistical Mechanical Ensembles
111111 is less likely to
occur, in a random se-
quence of 6-digit numbers,
than the number 845192.
In a random (uniformly dis-
tributed) sample, all num-
bers are equally likely to
occur, by definition. For mo-
Figure 5.4 Two microstates in an isolated system.
lecular systems, it is not
hard to argue that any spe-
cific set of positions and velocities of N particles in a volume V that has a given to-
tal energy, should be equally probable as any other specific set. There are, of
course, a lot more states that look like the right-hand side of Fig. 5.4 relative to
the left-hand side, the same way that there are a lot more 6-digit numbers with
non-identical digits than there are with all digits the same. The statement that all
microstates of a given energy are equally probable cannot be proved for the gen-
eral case of interacting systems. Thus, we adopt it as the basic Postulate of statisti-
cal mechanics:
Basic Postulate
of Statistical
Mechanics
For an isolated system at constant U, V, and N, all microscopic states of a system
are equally likely at thermodynamic equilibrium.
Just as was the case for the postulates of classical thermodynamics, the justification
for this statement is that predictions using the tools of statistical mechanics that rely
on this postulate are in excellent agreement with experimental observations for
many diverse systems, provided that the systems are ergodic.
As already suggested in 3.6, given that (U,V,N) is the number of microstates
with energy U, the probability of microstate in the microcanonical ensemble, ac-
cording to the postulate above, is:
1
P =
(U ,V ,N)
!
(5.4)
The
function
(U,V,N)
is
called
the
density
of
states,
and
is
directly
linked
to
the
entropy,
via
Botzmanns
entropy
formula,
S = kB ln .
It
is
also
related
to
the
fun-
!
damental
equation
in
the
entropy
representation,
with
natural
variables
(U,V,N).
Writing
the
differential
form
of
the
fundamental
equation
for
a
one-component
system
in
terms
of
,
we
obtain:
!
dS
= d ln = dU + P dV dN
kB
9/17/12 version
(5.5)
127
5.3 Microcanonical Ensemble: Constant U, V, and N
In
Eq.
5.5,
we
have
introduced
for
the
first
time
the
shorthand
notation
1/(kBT ) .
This
combination
appears
frequently
in
statistical
mechanics,
and
is
!
usually
called
the
inverse
temperature,
even
though
strictly
speaking
it
has
units
of
inverse
energy
[J1].
Differentiation
of
Eq.
5.5
provides
expressions
for
the
in-
verse
temperature,
pressure,
and
chemical
potential,
in
terms
of
derivatives
of
the
logarithm
of
the
number
of
microstates
with
respect
to
appropriate
variables:
ln
U
(5.6)
V ,N
ln
V = P
U ,N
!
(5.7)
ln
N =
U ,V
!
(5.8)
Example 5.1 A system with two states and negative temperatures
Consider a system of N distinguishable particles at fixed positions, each of which
can exist either in a ground state of energy 0, or in an excited state of energy . The
system is similar to that depicted in Fig. 3.13, except it has only 2 (rather than 3)
energy levels. Assuming that there are no interactions between particles, derive
expressions for the density of states and the temperature as a function of the ener-
gy, at the thermodynamic limit, N.
For a given total energy !U = M , the number of possible states is given by the ways
one can pick M objects out of N total particles:
N!
(U) = N =
M!(N M)!
M
!
At
the
limit
of
large
N,
we
can
use
Stirlings
approximation,
!ln(N!) N lnN N :
ln (U) = N lnN N M lnM + M (N M)ln(N M)+ N M
N
!
ln (U) = N lnN M lnM (N M)ln(N M)
!
The temperature as a function of U is obtained from Eq. 5.6, taking into account
that the volume V is not a relevant variable for this system:
ln
=
=
U N
ln
(M )
N
9/17/12 version
128
Chapter 5. Statistical Mechanical Ensembles
1
1
=
( N ln N M ln M (N M )ln(N M )) = ( ln M 1+ ln(N M ) + 1)
M
N
= ln
kT
NM
N
= ln 1
B =
M
M
!
1
N
ln 1
M
The
possible
values
of
the
normalized
energy
ratio,
U /Umax = M / N ,
range
from
0
!
(every
particle
is
in
the
ground
state)
to
1
(every
particle
is
in
the
excited
state).
The
relationship
between
M/N
and
the
temperature
is
shown
in
Fig.
5.5.
Low
val-
ues
of
M/N
correspond
to
low
temperatures.
Remarkably,
the
temperature
ap-
proaches
+
as
M/N
from
below,
and
then
returns
from
negative
infinity
to
just
below
zero
as
M/N
1.
Figure
5.5
Temperature
versus
normalized
energy
for
the
system
of
Example
Error!
Reference
source
not
found..
Do
these
results
make
sense?
Can
negative
temperatures
exist
in
nature?
It
turns
out
that
the
existence
of
negative
temperatures
is
entirely
consistent
with
thermo-
dynamics.
The
system
of
this
example
has
a
density
of
states
that
is
a
decreasing
function
of
the
total
energy
U
when
more
than
half
the
particles
are
in
the
excited
state.
Most
physical
systems
(e.g.
molecules
free
to
translate
in
space)
have
a
mon-
otonic
increase
in
the
number
of
states
at
higher
energies,
and
thus
cannot
exist
at
negative
temperatures;
however,
some
spin
systems
can
closely
approximate
the
system
of
this
example.
One
needs
to
realize
that
negative
temperatures
are
effec-
tively
higher
than
all
positive
temperatures,
as
energy
will
flow
in
the
negative
positive
direction
on
contact
between
two
systems
of
opposite
sides
of
the
dashed
line
M/N
=
,
corresponding
to
=
0,
or
T
=
.
9/17/12 version
5.3 Microcanonical Ensemble: Constant U, V, and N
129
5.4 Canonical Ensemble: Constant N, V, and T
Just as in macroscopic thermodynamics, in statistical mechanics we are interested
in developing relationships for systems subject to constraints other than constant
U, V, and N. In Chapter 4, a change of variables was performed through the Legen-
dre transform formalism, which will turn out to be highly relevant here as well.
The first example to consider will be a system of fixed volume V and number of
particles N, in thermal contact with a much larger reservoir, as shown in Figure
5.6. Because energy can be transferred between the small system and the reservoir
without a significant change in the reservoirs properties, the small system is effec-
tively at constant temperature, that of the reservoir. The set of microstates com-
patible with constant-NVT conditions is called the canonical ensemble. Going from
the microcanonical (UVN) to the canonical (NVT) ensemble is akin to taking the
first Legendre transformation of the fundamental equation in the entropy represen-
tation. Note that the order of variables (UVN TVN) is important when performing
Legendre transformations; however, convention dictates that the canonical en-
semble is referred to as the NVT ensemble the ordering of variables is unim-
portant once a given transformation has been performed.
How do we derive the relative
probabilities of microstates for
the constant-temperature small
system? The total system (small
system + reservoir) is under con-
stant-UVN conditions. In the pre-
vious sections of this chapter, we
suggested that all microstates
of the total system, which is at
constant energy, volume and
number of particles, are equally
probable. However, a given mi-
crostate of the small system
with energy U is consistent with
Figure 5.6 A small system in contact with a large
many possible microstates of the
reservoir.
reservoir the only constraint is
that the energy of the reservoir is UR = U U . The number of such microstates for the
reservoir is
(U ) = R (U U )
! R R
The
probability
of
finding
the
small
system
in
state
is
proportional
to
the
number
of
such
microstates,
9/17/12 version
130
Chapter 5. Statistical Mechanical Ensembles
P R (U U ) = exp ln R (U U )
!
(5.9)
We can Taylor-expand lnR around R (U) given that U is much smaller than U:
lnR
ln R (U U ) = ln R (U) U
+ !
(5.10)
U
!
Substituting
5.10
back
in
Eq.
5.9
and
using
Eq.
5.6,
we
can
incorporate
the
term
involving
Res
(that
does
not
depend
on
the
microstate
)
into
the
constant
of
proportionality
for
P :
P exp U
at
constant
N V T
!
(5.11)
This
is
a
very
important
result.
The
probability
of
each
microstate
in
the
ca-
nonical
ensemble
(constant
NVT)
decreases
exponentially
for
higher
energies.
The
probability
distribution
of
Eq.
5.11
is
known
as
the
Boltzmann
distribution.
In
order
to
find
the
absolute
probability
of
each
microstate,
we
need
to
nor-
malize
the
probabilities
so
that
their
sum
is
1.
The
normalization
constant
is
called
the
canonical
partition
function,
Q
and
is
obtained
from
a
summation
over
all
mi-
crostates,
canonical
partition
function
Q (N ,V ,T ) =
exp U
all!microstates!!
!
(5.12)
The probability of each microstate can now be written explicitly as an equality:
exp U
P =
at
constant
N V T
(5.13)
Q
!
The
probability
of
all
microstates
with
a
given
energy
U
is
the
a
sum
of
(U )
equal
terms,
each
at
the
volume
V
and
number
of
molecules
N
of
the
system:
(U)exp U
P(U) =
at
constant
N V T
(5.14)
Q
!
An
important
implication
of
these
derivations
is
that
the
energy
of
a
system
at
constant
temperature
is
strictly
fixed.
Instead
it
fluctuates
as
the
system
samples
different
microstates.
This
is
in
direct
contrast
with
the
postulate
of
classical
ther-
modynamics
that
three
independent
variables
(N,
V,
and
T
in
this
case)
fully
char-
acterize
the
state
of
a
system,
including
its
energy.
As
will
be
analyzed
in
detail
in
the
following
chapter,
fluctuations
of
quantities
such
as
the
energy
are
present
in
all
finite
systems,
but
their
relative
magnitude
decreases
with
increasing
system
size.
For
macroscopic
systems
fluctuations
are
negligible
for
all
practical
purposes,
except
near
critical
points.
In
any
statistical
mechanical
ensemble,
however,
we
9/17/12 version
131
5.4 Canonical Ensemble: Constant N, V, and T
need
to
make
a
clear
distinction
between
quantities
that
are
strictly
constant
(con-
strains,
or
independent
variables
in
the
language
of
Legendre
transformations),
and
those
that
fluctuate
(derivatives).
Any
thermodynamic
property
B
can
be
obtained
from
a
summation
over
mi-
crostates
of
the
value
of
the
property
at
a
given
microstate
times
the
probability
of
observing
the
microstate:
<B > = B P for any ensemble
(5.15)
all!microstates!
!
For example, the ensemble average energy !< U > in the canonical ensemble is
given by:
1
< U > = U exp U at
constant
N V T
(5.16)
Q !
!
Let
us
calculate
the
temperature
derivative
of
the
canonical
partition
function
Q
.
We
have:
U exp U
lnQ
1 exp(U )
=
=
= < U >
(5.17)
The
fundamental
equation
for
S/kB
,
Eq.
5.5,
has
U,
V
and
N
as
its
variables
and
,
P
,
and
as
its
derivatives.
Its
first
Legendre
transform
with
respect
to
U
is:
S
S
U
U TS
U =
=
= A
(5.18)
kB
kB kBT
kBT
!
This
is
a
function
of
,
V
,
and
N,
with:
(A)
(5.19)
= U
V ,N
!
Comparing
Eqs.
5.17
and
5.19,
we
see
that
the
former
(obtained
from
statisti-
cal
mechanics)
gives
the
ensemble
average
energy,
recognizing
that
the
energy
fluctuates
under
constant-NVT
conditions.
The
latter
expression,
obtained
from
thermodynamics
using
Legendre
transformations,
does
not
involve
averages
of
fluctuating
quantities.
At
the
thermodynamic
limit,
N
,
we
can
set
the
two
ex-
pressions
equal
,
and
obtain
a
direct
connection
between
the
canonical
partition
function
and
the
first
Legendre
transform
of
S /kB ,
!A = lnQ
(5.20)
Eq. 5.20 relates a thermodynamic quantity, the Helmholtz energy A, to a micro-
scopic one, the partition function Q. This also allows us to confirm the Gibbs en-
9/17/12 version
132
Chapter 5. Statistical Mechanical Ensembles
tropy
formula
(Eq.
3.20,
p.
59)
for
the
case
of
a
system
at
constant-N V T ,
in
the
thermodynamic
limit
N
:
= lnQ + U =
A +U S
=
kBT
kB
P lnP =
P lnQ U = lnQ P + PU =
Eq.!5.14
(5.21)
!
Given
that
lnQ
is
the
first
Legendre
transformation
of
ln,
we
can
now
ex-
!
press
all
the
first
derivatives
of
the
canonical
partition
function
Q ,
analogous
to
Eqs.
5.65.8
for
the
derivatives
of
the
microcanonical
partition
function
:
lnQ
= U
(5.22)
V ,N
!
lnQ
(5.23)
V = P
T ,N
!
lnQ
(5.24)
N =
T ,V
!
These
expressions
are
strictly
true
only
in
the
thermodynamic
limit
N;
for
finite
systems,
for
which
we
need
to
preserve
the
distinction
between
fluctuating
and
strictly
constant
quantities,
the
proper
expressions
involve
ensemble
averag-
es;
for
example,
the
correct
version
of
Eq.
5.22
is:
lnQ
= < U >
V ,N
!
(5.25)
Example 5.2 A system with two states in the NVT ensemble
Consider the system of N distinguishable particles at fixed positions, each of which
can exist either in a ground state of energy 0, or in an excited state of energy , in-
troduced in Example 5.1. Determine the mean energy <U> as a function of temper-
ature in the canonical ensemble, and compare the result to the microcanonical en-
semble calculation of Example 5.1.
We denote the state of each particle i = 1,2, N by a variable li which can take the
values 0 or 1, denoting the ground or excited state. The total energy is
N
U = li
i=1
!
The
partition
function
in
the
canonical
ensemble
is:
9/17/12 version
5.4 Canonical Ensemble: Constant N, V, and T
lnQ = ln e
Q =
133
N
N
exp
l
=
exp(li )
i
i=1 l1 ,l2 ,...,lN =0,1 i=1
l1 ,l2 ,...,lN =0,1
Now
we
can
use
a
mathematical
identity
that
will
turn
out
to
be
useful
in
all
cases
in
which
a
partition
function
has
contributions
from
many
independent
particles.
The
sum
contains
2N
terms,
each
consisting
of
a
product
of
N
exponentials.
We
can
regroup
the
terms
in
a
different
way,
in
effect
switching
the
order
of
the
summa-
tion
and
multiplication:
!l1 ,l2 ,...,lN =0,1
exp(li ) = e
i=1
li
= (1+ e )N
i=1 li =0,1
You
can
easily
confirm
that
the
switched
product
contains
the
same
2N
terms
as
before.
The
final
result
for
the
partition
function
is:
!lnQ = N ln(1+ e )
The
ensemble
average
energy
<U>
is
lnQ
e
N
<U > =
=
N
=
() N ,V
1+ e 1+ e
!
The
microcanonical
ensemble
result
can
be
written
as:
N
N
N
ln 1 =
= 1 = e M =
M = U =
M
M kBT
1+
e
1+
e
!
The
only
difference
is
that
in
the
canonical
ensemble
the
energy
is
a
fluctuating
(rather
than
a
fixed)
quantity.
Note
also
that
the
canonical
ensemble
derivation
does
not
entail
any
approximations;
the
same
result
is
valid
for
any
N.
By
contrast,
the
microcanonical
energy
was
obtained
through
the
use
of
Stirlings
approxima-
tion,
valid
as
N.
Small
differences
between
the
two
ensembles
are
present
for
finite
N.
5.5 Generalized Ensembles and Legendre Transforms
The previous two sections illustrated how, starting from the fundamental equation
in the entropy representation and Boltzmanns entropy formula, one can obtain
relationships between macroscopic and microscopic quantities, first in the UVN
(microcanonical) ensemble, with key quantity the density of states, . A first Le-
gendre transformation of U to T resulted in the canonical ensemble, with partition
function Q. This process can be readily generalized to obtain relationships be-
tween microscopic and macroscopic quantities and partition functions for a sys-
tem under arbitrary constraints.
9/17/12 version
134
Chapter 5. Statistical Mechanical Ensembles
In our derivation, we start from the multicomponent version of the fundamen-
tal equation in the entropy representation,
n
dS
= d ln = dU + PdV i dNi
kB
i=1
(5.26)
We have already seen the first Legendre transformation:
S
y (1) = U = (TS U) = A
kB
!
(5.27)
The
relationships
between
the
original
function
and
the
first
transform
are
de-
picted
in
the
table
below.
y(0)
=
S/kB
=
ln
y(1) = A/kBT = lnQ
Variable
Derivative
Variable
Derivative
1/(kBT) =
1/(kBT) =
P/(kBT) = P
P/(kBT) = P
Ni
i/(kBT) = i
Ni
i/(kBT) = i
These
relationships
link
microscopic
to
macroscopic
properties
and
are
strictly
valid
at
the
thermodynamic
limit,
N.
One
should
keep
in
mind
that
in
each
en-
semble,
the
variables
of
the
corresponding
transform
are
strictly
held
constant,
defining
the
external
constraints
on
a
system,
while
the
derivatives
fluctuate
they
take
different
values,
in
principle,
for
each
microstate
of
the
ensemble.
The
magni-
tude
of
fluctuations,
and
connections
to
thermodynamic
derivatives,
will
be
made
in
Chapter
6.
Probabilities
of
microstates
in
two
statistical
ensembles
have
already
been
de-
rived.
Microstate
probabilities
are
all
equal
in
the
microcanonical
ensemble,
from
the
basic
postulate
of
statistical
mechanics;
they
are
equal
to
the
Boltzmann
factor,
exp(U),
normalized
by
the
partition
function
for
the
canonical
ensemble
(Eq.
5.13).
Note
that
the
factor
U
that
appears
in
the
exponential
for
microstate
probabilities
in
the
canonical
ensemble
is
exactly
equal
to
the
difference
between
the
basis
function
and
its
first
Legendre
transform,
x11 .
!
One
can
continue
this
with
Legendre
transforms
of
higher
order.
The
probabil-
ities
of
microstates
in
the
corresponding
ensembles
can
be
derived
in
a
way
com-
pletely
analogous
to
the
derivation
for
the
canonical
ensemble,
involving
a
subsys-
9/17/12 version
135
5.5 Generalized Ensembles and Legendre Transforms
tem
and
bath
of
constant
temperature,
pressure,
chemical
potential,
etc.
In
general,
the
kth
transform
of
the
basis
function
is:
S
S
y (0) = !!!!!;!!!!!!!!!!y (k ) = 1x1 2x2 kxk
kB
kB
!
(5.28)
where
i
is
the
i-th
derivative
of
y(0)
with
respect
to
variable xi .
The
probability
!
!
of
a
microstate
in
the
ensemble
corresponding
to
the
kth
transform
is
given
by
P exp 1x1 2x2 kxk
(5.29)
!
where the variables xi and derivatives i refer to the original function y (0) . The
!
!
!
normalization factor (partition function) of the ensemble corresponding to the
transformed function, y (k ) is:
!
=
(5.30)
exp 1x1 2x2 kxk
all!microstates!
!
Using the partition function , the probability P can be written as an equality:
P =
!
exp 1x1 2x2 kxk
(5.31)
As
was
the
case
for
the
canonical
ensemble,
the
partition
function
is
simply
re-
lated
to
the
transformed
function,
y (k ) :
!
ln = y (k )
!
(5.32)
Example 5.3 Gibbs Entropy Formula
The Gibbs entropy formula,
S = kB P lnP
!
was
derived
in
3.6
for
the
microcanonical
ensemble.
Show
that
this
relationship
is
valid
for
all
statistical
ensembles.
We
use
the
expression
for
the
probability
of
microstates,
P ,
in
a
generalized
en-
semble,
Eq.
5.31:
P lnP = P ln
exp 1x1 2x2 kxk
)=
P ( 1x1 2x2 kxk ln)
Now
recall
that
the
variables
for
the
k-th
transform
are
1 ,2 ,, k ,xk+1 ,xn+2 ,
!
which
are
strictly
constant
in
the
corresponding
ensemble,
while
the
derivatives,
x ,x ,,xk , k+1 , n+2 fluctuate.
We
can
rewrite
the
equation
above
taking
this
! 1 2
into
account:
9/17/12 version
136
Chapter 5. Statistical Mechanical Ensembles
P ( 1x1 2x2 kxk ln)
= 1 Px1 2 Px2 k Pxk ln =
= 1 < x1 > 2 < x2 > k < xk > ln
From Eqs. 5.28 and 5.32,
S
ln = y (k ) = 1x1 2x2 kxk
kB
!
At
the
thermodynamic
limit,
N,
so
there
is
no
distinction
between
ensemble
averages
and
thermodynamic
properties,
< xi > xi .
Replacing
!ln and
simplify-
!
ing,
P lnP = k
, QED
Example
5.4
Grand
Canonical
(VT)
(NPT)
Ensemble
The
grand
canonical
(constantVT)
and
constant-pressure
(NPT)
ensembles
are
frequently
used
in
computer
simulations.
Derive
the
partition
functions,
probabil-
ity
of
microstates,
and
derivative
relationships
in
these
two
ensembles
for
a
1-
component
system.
Starting
from
the
fundamental
equation
in
the
entropy
representation
with
order-
ing
of
variables
y (0) = S(U ,N ,V )/kB = ln ,
the
grand
canonical
ensemble
partition
!
function
corresponds
to:
S
TS U + N
ln = y (2) = U + N =
kB
kBT
!
The
microstates
possible
in
this
ensemble
include
all
possible
particle
numbers
from
0
to
,
and
all
possible
energy
levels.
The
Euler-integrated
form
of
the
fundamental
equation
is
!U = TS PV + N ,
so
that
the
partition
function
of
the
grand
canonical
ensemble
can
be
linked
to
the
follow-
ing
thermodynamic
property
combination:
PV
ln =
= PV
kBT
!
9/17/12 version
137
5.5 Generalized Ensembles and Legendre Transforms
y (2) = PV = ln
!
y (0) = S /kB = ln
!
Variable
Derivative
Variable
Derivative
U
N
V
U
N
P
For
example,
the
average
number
of
molecules
in
the
system
is
given
by:
ln
ln
= < N >
() = < N > kBT
,V
,V
!
The
probability
of
microstates
in
this
ensemble
is:
P =
!
exp(U + N )
, where = exp(U + N )
Example
5.5
Constant-pressure
(NPT)
Ensemble
The
constant-pressure
(NPT)
ensemble
is
also
frequently
used
in
computer
simu-
lations.
Derive
the
partition
functions,
probability
of
microstates,
and
derivative
relationships
in
this
ensembles
for
a
1-component
system.
Ee
start
from
from
the
fundamental
equation
in
the
entropy
representation
with
ordering
of
variables
y (0) = S(U ,V ,N)/kB ,
and
obtain
the
second
transform:
!
S
TS U PV
ln = y (2) = U PV =
= N = G
kB
kBT
!
The
microstates
possible
in
this
ensemble
include
all
possible
volumes
from
0
to
,
and
all
possible
energy
levels.
The
derivative
table
is:
y (2) = N = ln
!
y (0) = S /kB = ln
!
Variable
Derivative
Variable
Derivative
U
V
N
P
N
U
V
9/17/12 version
138
Chapter 5. Statistical Mechanical Ensembles
For example, the average volume is given by:
ln
ln
= < V >
(P) = < V > kBT P
,N
,N
!
The
probability
of
microstates
in
this
ensemble
is:
P =
!
exp(U PV )
, where = exp(U PV )
9/17/12 version
Chapter
6.
Equilibrium,
Stability,
and
Fluctuations
The
First
and
Second
Laws
of
Thermodynamics
allow
us
to
make
powerful
general
statements
about
the
overall
behavior
of
systems
in
thermodynamic
equilibrium
with
respect
to
external
interactions
in
particular,
we
have
learned
how
to
obtain
work
and
heat
flows
and
entropy
changes
of
a
system
and
its
environment.
We
have
also
established
that
irreversible
(spontaneous)
changes
within
a
system
re-
sult
in
a
net
increase
in
the
entropy
of
the
universe.
We
have
not
yet
examined,
however,
the
detailed
internal
requirements
and
constraints
imposed
by
thermo-
dynamic
equilibrium
on
properties
and
thermodynamic
functions
of
systems.
Ex-
perience
suggests
that
for
a
system
at
equilibrium,
density
does
not
need
to
be
uni-
form;
for
example,
a
vessel
containing
a
liquid
and
its
saturated
vapor
clearly
has
density
variations.
However,
temperature
is
uniform
throughout
systems
at
equi-
librium,
at
least
at
the
level
of
macroscopic
measurements.
This
must
somehow
be
connected
to
the
overall
condition
imposed
by
the
Second
Law
that
the
entropy
of
the
universe
is
maximized
at
equilibrium,
but
the
link
has
not
yet
been
made
ex-
plicit.
A
key
difference
between
macroscopic
thermodynamic
and
statistical
mechanical
points
of
view
is
that
the
latter
recognizes
the
existence
of
fluctuations
occuring
in
small
regions
of
a
system
at
equilibrium.
Such
fluctuations
are
completely
absent
in
macroscopic
thermodynamics.
We
have
alluded
in
the
previous
chapter
that
the
two
seemingly
contradictory
statements
are
reconciled
at
the
limit
N.
In
the
present
chapter,
we
will
develop
the
necessary
framework
for
performing
this
reconciliation.
Interestingly,
the
existence
of
microscopic
fluctuations
will
turn
out
to
be
intimately
linked
to
thermodynamic
stability:
macroscopic
systems
become
unstable
when
their
microscopic
fluctuations
become
unbounded.
The
goals
of
the
present
chapter
are
to
examine
the
conditions
of
equilibrium
and
stability
of
thermodynamic
systems,
and
to
obtain
relationships
expressing
microscopic
fluctuations
in
terms
of
system
properties.
We
also
obtain
constraints
on
the
signs
and
relative
magnitudes
of
thermodynamic
derivatives
that
follow
from
the
equilibrium
and
stability
conditions.
In
Chapter
5,
we
studied
methods
to
Draft
material
from
Statistical
Thermodynamics
2012,
A.
Z.
Panagiotopoulos
10/4/12 version
152
153
6.1 Equilibrium Criteria
manipulate derivatives of fundamental equations and to obtain relationships
among them. Some of these derivatives, e.g. the thermal expansion coefficient P ,
can be positive for some systems and negative for others. A well-known example
of negative thermal expansion coefficient is liquid water between 0 and 4 C. With-
in this temperature range, water contracts upon heating, while most other sub-
stances (as well as water outside this range) expand on heating. Does thermody-
namics place any restrictions on derivatives such as the isothermal compressibility
or heat capacities CV and CP? These and similar questions are addressed in the
sections that follow. Conditions of equilibrium also allow us to establish general
constraints on how many phases can coexist at equilibrium (Gibbs phase rule).
6.1 Equilibrium Criteria
One of the consequences of the Second Law of Thermodynamics is that entropy is
maximized for isolated closed systems at equilibrium (see 3.4). Isolated systems
can be considered to be at constant (U ,V ,N ), since they do not exchange heat,
work or mass with their environment. One example of an isolated system is the
universe itself. Irreversible (spontaneous) processes bring systems from off-
equilibrium states to equilibrium states. We have shown (Eq. 3.10, p. 45) that the
entropy of an isolated closed system increases when the system undergoes an ir-
reversible process leading to an equilibrium state. Therefore,
S is maximized at equilibrium when ( U ,V ,N ) are constant
This
condition
turns
out
to
be
a
good
starting
point
for
deriving
equilibrium
criteria
for
systems
subject
to
different
constraints.
Let
us
consider
a
small
pertur-
bation
within
an
isolated
system
containing
a
pure
component,
as
shown
in
Fig.
6.1.
This
perturbation
could
be
a
change
in
energy,
volume
or
amount
of
material
in
a
region
that
is
labeled
system
I,
while
the
rest
of
the
isolated
system
is
labeled
system
II.
The
first-order
variation
in
the
overall
entropy
is:
S = SI + SII
(6.1)
!
Using
the
fundamental
equation
in
the
entropy
representation
for
a
one-compo-
nent
system
(p.
82):
P
1
SI = UI + I VI I NI
TI
TI
TI
(6.2)
PII
II
1
SII = UII + VII NII
TII
TII
TII
!
10/4/12 version
154
Chapter 6. Equilibrium, Stability, and Fluctuations
Since
the
overall
system
is
isolated,
its
energy,
volume,
and
number
of
moles
are
fixed;
variations
within
the
two
subsystems
must
be
linked
so
as
to
satisfy:
UI + UII = 0 UI = UII
VI + VII = 0! VI = VII
(6.3)
N + NII = 0 NI = NII
! I
Substituting
the
expressions
from
Eqs.
6.2
and
6.3
into
Eq.
6.1,
we
obtain:
1 1
P P
S = UI + I II VI I II NI
TI TII
TI TII
TI TII
!
(6.4)
The variations UI , VI , and NI are independent of each other: we can certain-
ly envision a fluctuation in the volume of a subsystem that leaves its energy and
number of moles unchanged. The fact that the entropy of the overall isolated sys-
tem is a maximum at equilibrium implies that all the prefactors of the independent
variations in Eq. 6.4 must be zero:
!
equilibrium
criteria,
1-component
systems
P P
I II
1 1
= 0!!!; I II = 0!!!;
= 0
TI TII
TI TII
TI TII
TI = TII
PI = PII
(6.5)
= II
! I
In
words,
maximization
of
the
entropy
implies
that
temperature,
pressure
and
chemical
potential
must
be
uniform
at
equilibrium.
Even
though
we
derived
this
Figure 6.1 A subsystem within an isolated system.
10/4/12 version
155
6.1 Equilibrium Criteria
result for the case of an isolated system at
constant (U ,V ,N ), it applies to all sys-
tems at equilibrium, irrespective of the
constraints placed upon them. This is be-
cause setting a different constraint, e.g.,
constant (T ,V ,N ), imposes uniformity of
temperature throughout the total system,
so the first condition of Eq. 6.5 is auto-
matically satisfied. Generalization to n-
component systems is straightforward,
resulting in the condition that the chemi-
cal potential for each component must be
uniform in multicomponent systems at
equilibrium, in addition to the tem-
perature and pressure equality condi-
tions:
Figure
6.2
Variation
of
entropy
for
a
system
at
constant
U.
TI = TII
PI = PII
(6.6)
= i ,II !!for!i = 1,2,...n
! i ,I
Entropy is the fundamental equation with natural variables (U,V,N). The fact
that it is a maximum for an isolated system when variables (U,V,N) are held con-
stant suggests the hypothesis that other fundamental equations also reach an ex-
tremum under the constraint that their own natural variables are held fixed. To
show this, we first turn our attention to the energy U, which has natural variables
(S ,V ,N ). As we will immediately show, maximization of entropy S at constant
(U ,V ,N ) implies minimization of the energy U at constant ( S ,V ,N ).
Consider Fig. 6.2, showing schematically the variation of entropy as a system
moves slightly away from equilibrium point A at constant (U ,V ,N ). Since entropy
is a maximum at equilibrium, the off-equilibrium state of the system (point B, with
UB = UA) will have entropy lower than that of the equilibrium state A. Now, we
would like to consider a similar small excursion away from the original equilibri-
um state A of the system, but this time at constant S. We can bring the system from
point B to point C, which has the same value of the entropy as the initial state A, by
supplying a small amount of heat, Q, in a reversible fashion, so that Q = TS. Since
S = SA SB = SC SB > 0 because the entropy is a maximum at equilibrium, the amount
of heat is also positive, Q > 0; heat must be supplied to the system. We perform a
First Law balance taking into account that work is zero because of the constant-
volume condition:
10/4/12 version
equilibrium
criteria,
n-component
systems
156
Chapter 6. Equilibrium, Stability, and Fluctuations
Figure
6.3
System
in
contact
with
a
thermal
reservoir
U = UC UB = UC U A = Q +
> 0
V !is!constant
!
The
small
change
from
an
equilibrium
to
an
off-equilibrium
state
resulted
in
U
>
0
at
constant
entropy:
the
energy
increases
when
we
move
away
from
equilib-
rium
at
constant
( S ,V ,N ).
Therefore,
we
have
shown
that:
U is minimized at equilibrium when ( S ,V ,N ) are constant
The
next
step
is
to
examine
the
behavior
of
systems
that
are
subject
to
con-
straints
different
from
(U ,V ,N )
or
(S ,V ,N ).
The
derivations
parallel
the
develop-
ment
of
alternate
representations
of
the
fundamental
equation
in
Chapter
5,
in
which
we
moved
from
the
energy
U (S ,V ,N )
to
the
Helmholtz
energy
A (T ,V ,N )
or
Gibbs
free
energy
G (T ,P ,N ).
First,
let
us
consider
the
constant-(T ,V ,N )
case
by
analyzing
a
small
system
in
contact
with
a
large
thermal
reservoir
as
shown
in
Fig.
6.3.
Changes
in
energy
of
the
small
system
result
in
negligible
changes
in
the
prop-
erties
of
the
large
reservoir,
which
imposes
its
temperature
on
the
small
system.
Water
baths
used
in
chemistry
and
biology
labs
are
good
examples
of
(approxi-
mately)
constant-temperature
reservoirs.
We
will
consider
the
sum
of
(system
+
reservoir)
to
constitute
a
total
system
that
is
isolated
(constant
U ,V ,N ),
so
that:
Stotal = S + SR
10/4/12 version
(6.7)
157
6.1 Equilibrium Criteria
We
have
used
the
subscripts
total
and
R
to
distinguish
properties
of
the
total
sys-
tem
and
the
reservoir,
respectively.
The
system
of
interest
(the
one
being
held
at
constant
T,
V
and
N),
is
assigned
no
special
label.
For
the
total
system,
the
entropy
is
a
maximum
at
equilibrium
as
the
system
is
at
constant
( U ,V ,N ).
For
any
process
that
takes
the
system
from
an
off-equilibrium
to
an
equilibrium
state,
we
must
have
Stotal > 0 .
For
the
total
system,
we
have:
U
= 0 U + UR = U +TSR
! total
(6.8)
Here,
we
have
assumed
that
the
transfer
of
heat
from
the
system
to
the
reservoir
takes
place
reversibly,
since
both
system
and
reservoir
are
at
the
same
tempera-
ture.
Furthermore,
there
is
no
volume
change
or
work
associated
with
the
reser-
voir,
so
UR
=
TSR.
Now,
from
Eq.
6.7,
we
have:
S = Stotal S
! R
Eq.
6.8
now
gives:
U
= 0 = U TS +TStotal U TS = TStotal < 0
! total
Since the system is at constant T,
!U TS = (U TS) = A < 0
We
have
thus
shown
that
moving
towards
equilibrium
results
in
lowering
of
the
Helmholtz
free
energy
A,
so
that:
A is minimized at equilibrium when ( T ,V ,N ) are constant
The energy U is not minimized at equilibrium at constant (T ,V ,N ), except at
very low absolute temperatures, for which the Helmholtz energy A = U TS becomes
identical to U. One manifestation of this fact is that solids melt above a specific
temperature, even though the lowest energy arrangement of molecules of most
substances corresponds to a crystalline solid. The resulting liquids have higher
energy U than their crystals, but also higher entropy S. The Helmholtz energy A is
lower for the liquids above the melting point, making them the stable thermody-
namic states. In other words, liquids exist because A is minimized rather than U at
constant temperature. Another example is the spontaneous folding of many pro-
teins in solution into specific three-dimensional patterns encoded in their amino
acid sequences. Even though energy minimization is often used to search for stable
folded states, the correct thermodynamic function to be minimized is A, not U.
However, calculation of A is much harder than calculation of U, so minimization of
A is often not done because it is computationally impractical.
10/4/12 version
158
Chapter 6. Equilibrium, Stability, and Fluctuations
Let us now consider closed systems at constant T, P and N. A schematic dia-
gram of such a system is shown in Fig. 6.4. The small system of interest is connect-
ed on the left side to a large thermal reservoir by a diathermal rigid wall and on
the right side to a large volume reservoir by a movable, insulated piston. The
thermal reservoir imposes its temperature, and the volume reservoir imposes its
pressure on the system of interest, since variations in the systems energy or vol-
ume are too small to affect the properties of the reservoirs. Once more, we will
consider the sum of (system + thermal reservoir + volume reservoir) to constitute
a total system that is isolated (constant U ,V ,N ), so that its entropy is maximized
at equilibrium:
S
= S + ST'res + SP'res > 0
! total
(6.9)
The
entropy
change
of
the
volume
reservoir
is
zero
because
there
is
no
heat
flow
into
or
out
of
it.
The
total
energy
Utot
is
constant,
so
that:
U total = 0 = U + UT(res + UP(res = U +TST(res P VP(res
VP(res =V
U
= 0 = U +TST(res + PV TST(res = U PV
! total
Substituting
in
Eq.
6.9:
TStotal = TS +TST'res = TS U PV = (U TS + PV ) = G > 0
!
G < 0
Figure 6.4 System in contact with thermal and volume reservoirs
10/4/12 version
159
6.1 Equilibrium Criteria
So
that:
G is minimized at equilibrium when ( T ,P ,N ) are constant
In
a
similar
fashion,
one
can
prove
that
for
any
set
of
constraints
on
a
system,
the
thermodynamic
function
that
corresponds
to
a
fundamental
equation
with
the
corresponding
variables
is
minimized
or
maximized
at
equilibrium,
depending
on
whether
we
start
from
the
fundamental
equation
in
the
energy
or
entropy
repre-
sentations.
A
connection
to
the
statistical
mechanical
ensembles
introduced
in
the
previ-
ous
chapter
can
also
be
made.
We
have
seen
that
a
given
set
of
external
constraints
imposed
on
a
system
define
a
statistical
ensemble,
with
partition
function
equal
to
the
exponential
of
the
corresponding
Legendre
transformation.
The
starting
point
is
y (0) = S /kB ,
which
corresponds
to
constantUVN
conditions.
The
condition
of
!
equilibrium
for
a
system
at
constantUVN,
is,
as
we
have
just
established,
the
max-
imization
of
the
entropy
S,
and
therefore
also
of
y (0) .
The
first
Legendre
transform
!
corresponds
to
a
system
at
constantNVT
conditions:
S
U
U TS
lnQ = y (1) =
=
= A
kB kBT
kBT
!
We
have
just
established
that
the
Helmholtz
free
energy
A
is
minimized
at
con-
stantNVT
conditions,
so
that
A
is
maximized.
Thus,
we
can
generalize
this
ob-
servation
as
follows.
At
the
set
of
conditions
that
define
a
statistical
mechanical
ensemble,
the
thermo-
dynamic
function
corresponding
to
the
partition
function
of
the
ensemble
is
max-
imized
at
equilibrium.
Example
6.1
Determine
the
thermodynamic
potential
functions
being
minimized
or
maximized
at
equilibrium:
(a)
at
constant
( S ,P ,N )
(b)
at
constant
( T ,V , )
(c) at constant (1/T ,P/T ,N)
For
(a),
we
can
see
directly
that
the
thermodynamic
function
being
minimized
is
the
fundamental
equation
in
the
variables
( S ,P ,N ):
H
(S,
P,
N)
=
U
+
PV
is
minimized
at
equilibrium
for
constant
(S,
P,
N)
10/4/12 version
160
Chapter 6. Equilibrium, Stability, and Fluctuations
For
(b),
the
function
being
minimized
is
a
Legendre
transform
of
U
that
we
have
not
formally
defined,
but
should
be
a
function
of
(T,
V,
).
To
obtain
this
function,
we
need
to
transform
T
and
N
in
U
(S,
V,
N),
so
that
the
function
is
=
UTS
=
PV
We
conclude
that
the
function
=
PV
is
minimized
at
equilibrium
for
constant
(T,
V,
),
given
that
the
starting
point
was
U,
so
that
P V
is
maximized.
For
(c),
the
variables
are
related
to
the
entropy
representation
of
the
fundamental
equation,
1
P
dS = dU + dV dN
T
T
T
!
To
obtain
a
function
of
(1/T ,P/T ,N),
we
need
to
transform
the
first
two
variables
to
obtain
the
function:
S
U/T
PV/T
=
(
U
TS
+PV
)/T
=
G/T,
which
is
maximized
at
equilibrium,
as
the
starting
point
was
S.
More
complex
types
of
equilibria
can
be
encountered
in
systems
with
internal
boundaries.
A
noteworthy
example
of
such
boundaries
is
semipermeable
mem-
branes
that
do
not
allow
exchange
of
molecules
of
certain
components
be-
tween
the
two
sides
of
the
membrane.
A
schematic
of
such
a
system
is
shown
in
Fig.
6.5.
The
overall
system
is
considered
under
conditions
of
isolation,
at
constant
U,
V,
and
N.
At
equilibrium,
the
entropy
of
the
total
system
is
maximized:
Figure
6.5
System
with
a
rigid
membrane
permeable
only
to
component
A.
S = SI + SII
! tot
(6.10)
Since
the
membrane
is
rigid,
the
volumes
of
the
two
compartments
are
constant;
the
number
of
moles
of
component
B
is
also
constant
on
both
sides;
however,
the
energies
can
fluctuate
because
of
molecules
of
component
A
moving
across
the
membrane.
The
variations
of
entropy
are:
SI =
A ,I
1
UI
N A ,I
TI
TI
A ,II
1
SII = UII
N A ,II
TII
TII
!
10/4/12
version
(6.11)
161
6.1 Equilibrium Criteria
The
conditions
of
isolation
of
the
total
system
impose
conservation
of
energy
and
number
of
moles
of
A
in
the
two
regions
UI + UII = 0 UI = UII
N + N A ,II = 0 N A ,I = N A ,II
! A ,I
(6.12)
Substituting in the expression for Stot above,
!
A ,I A ,II
1 1
Stot = UI
N
TI
TII A ,I
TI TII
!
At
equilibrium,
the
first-order
entropy
variation
must
vanish
for
all
possible
per-
turbations,
so
that
the
prefactors
to
UI and
N A ,I must
both
be
zero.
The
condi-
!
!
tions
of
equilibrium
are
then:
TI = TII
= A ,II
! A ,I
(6.13)
6.2 Stability Criteria
We have just established in 6.1 that equilibrium in thermodynamic systems un-
der a certain set of external constraints implies that a thermodynamic potential
(fundamental equation) is minimized or maximized. There are, however, addition-
al mathematical requirements for minimization or maximization of functions. For
a simple one-dimensional mechanical system of a ball in a gravitational well (Fig.
6.6), the energy function U is at a minimum with respect to position x when:
dU
= 0
[condition
of
extremum]
! dx
(6.14)
Eq. 6.14 is a necessary but not sufficient condition for minimization of U. In par-
Figure 6.6 Types of mechanical equilibrium in a one-dimensional system.
10/4/12 version
162
Chapter 6. Equilibrium, Stability, and Fluctuations
ticular,
the
following
additional
condition
must
be
satisfied
by
the
second
deriva-
tive
of
U
at
the
point
at
which
the
first
derivative
vanishes
to
make
it
a
minimum:
d 2U
! dx
> 0 [minimization condition]
(6.15)
The
left-most
panel
of
Fig.
6.6
satisfies
the
condition
of
Eq.
6.15
and
corre-
sponds
to
a
stable
equilibrium
state.
Small
perturbations
of
the
system
to
either
left
or
right
of
the
minimum
produce
a
restoring
force
that
returns
the
system
to
the
stable
equilibrium
point.
For
the
second
panel
from
the
left,
the
second
derivative
is
negative
and
the
point
corresponds
to
a
maximum.
Such
a
condition
corre-
sponds
to
unstable
equilibrium.
At
exactly
the
top
of
the
hill
the
forces
on
the
ball
are
zero,
so
in
principle
the
ball
is
at
equilibrium.
However,
the
slightest
perturba-
tion
away
from
this
point
to
either
left
or
right
results
in
forces
that
move
the
ball
away
from
the
point
of
equilibrium;
systems
cannot
be
kept
for
any
significant
amount
of
time
at
unstable
equilibrium,
but
move
away
towards
stable
equilibri-
um
states.
An
additional
possibility
for
a
type
of
equilibrium
state
is
illustrated
in
the
third
panel
of
Fig.
6.6.
The
well
in
this
case
has
two
minima,
a
shallow
one
to
the
left
and
a
deeper
one
to
the
right.
There
is
no
simple
mathematical
test
to
distin-
guish
between
a
global
minimum
and
a
local
minimum:
in
both
cases,
the
first
de-
rivative
vanishes
and
the
second
derivative
of
the
function
at
the
minimum
is
posi-
tive.
Systems
that
are
at
a
local
minimum
of
the
appropriate
thermodynamic
po-
tential
are
said
to
be
in
metastable
equilibrium.
Small
perturbations
away
from
a
metastable
equilibrium
state
return
to
the
starting
point,
but
large
perturbations
can
lead
to
a
drastically
different
equilibrium
point.
As
the
depth
of
the
metastable
well
is
reduced,
we
reach
a
limit
of
stability
(right-most
panel
of
Fig.
6.6),
for
which:
dU
d 2U
= 0!!!and!!!
= 0
[limit
of
stability]
dx 2
! dx
(6.16)
Real thermodynamic systems are frequently found in metastable states. A liq-
uid being heated in a clean glass container may exceed its boiling point in the ab-
sence of nucleation sites. Rapid boiling will occur when a nucleation event takes
place in such a superheated liquid, with possible risks to nearby people and equip-
ment. This is the reason boiling stones providing nucleation sites are always
used in chemistry laboratories when heating liquids. In a similar way, a liquid may
cool below its melting temperature without nucleation of the solid, becoming a
supercooled liquid, but provision of a suitable nucleus in such a system results in
rapid crystallization. Jet aircraft often leave behind contrails because their ex-
haust gases provide nucleation sites for supersaturated water vapor in the atmos-
phere.
10/4/12 version
163
6.2 Stability Criteria
One may well question the applicability of thermodynamics to such metasta-
ble states. After all, we have established in Chapter 1 that thermodynamics applies
only to stable equilibrium states, fully characterized by n+2 independent thermo-
dynamic variables. Metastable states are not, strictly speaking, at equilibrium.
However, if they are sufficiently long-lived, they are indistinguishable from stable
equilibrium states and their thermodynamic properties are well defined. Recalling
the discussion in 1.3 of time scales involved in deciding if a boundary is permea-
ble. There are similar considerations of time scales with respect to metastable
states. If the time scales of interest are much shorter than the metastability barrier
crossing times, metastable states can be properly described by equilibrium ther-
modynamics.
Up to this point, the conditions of equilibrium and stability, Eqs. 6.14 to 6.16,
have been written for one-dimensional functions. Thermodynamic potentials are
functions of n+2 variables, so the conditions of maximization or minimization are
more complex. Mathematically, the condition of stability for functions of many var-
iables is expressed as a condition on the sign of its second-order variation. For ex-
ample, in the case of a system at constant ( S ,V ,N ) the energy is minimized at equi-
librium, so for the first-order variation we have U = 0. For the second-order varia-
tion we must have:
2U > 0
2 U
(S)2 +
2
2 U
(V )2 +
2
2 U
2
(N)2 +
S
V
N
2
2
U
U
2 U
!!!!!!!!!!!!!!!!!!!!!!!! + 2
SV +
SN +
VN > 0
S N
V N
S V
Since the variables S, V, and N are independent, we can consider variations in
which only one of the S, V or N terms are non-zero. In those cases, only one of
the first three terms will contribute to 2U. To ensure that 2U > 0 in this case, a
necessary but not sufficient condition is that the corresponding coefficients must
be positive:
2U
T
T
T
>0
=
>0
> 0
2
CV
S V ,N NCV
S V ,N
!
(6.17)
2U
P
P
< 0 < 0
2 >0
V S ,N
V S
V S ,N
!
(6.18)
2U
2 >0
> 0
N S ,V
N S ,V
!
(6.19)
10/4/12 version
164
Chapter 6. Equilibrium, Stability, and Fluctuations
Eq.
6.17
is
known
as
the
condition
of
thermal
stability
and
suggests
that
the
constant-volume
heat
capacity
of
any
stable
thermodynamic
system
must
be
posi-
tive.
Even
though
we
derived
this
expression
for
a
one-component
system
at
con-
stant
( S ,V ,N ),
this
is
a
general
relationship
valid
also
for
multicomponent
systems
under
arbitrary
constraints.
Note
also
that
at
constant
N,
we
can
move
from
exten-
sive
to
intensive
variables
in
expressing
the
final
form
of
the
stability
criteria.
We
have
already
suggested
that
these
are
necessary
but
not
sufficient
conditions
of
stability
for
thermodynamic
systems.
In
order
to
obtain
necessary
and
sufficient
conditions,
we
need
to
convert
!2U to
a
quadratic
form.
We
can
do
this
as
follows.
We
start
from
U = y (0)
as
the
basis
function.
The
second-order
variation
of
U
can
be
!
written
as:
2U = 2y (0) =
!
n+2 n+2
yij(0) xix j
(6.20)
i=1 j=1
Now we define auxiliary variables, zk as follows:
!
zk = xk +
n+2
ykj(k )x j !!!!for!k = 1,2,,n +1
j=k+1
(6.21)
z
= xn+2
! n+2
It
can
then
be
shown
using
the
properties
of
Legendre
transformations
that:
2U = 2y (0) =
!
n+1
ykk(k1) zk2
(6.22)
k=1
In
other
words,
the
use
of
the
auxiliary
variables
zk reduces
the
fundamental
equa-
!
tion
for
U
to
a
quadratic
form.
We
demonstrate
in
the
example
below
how
this
ap-
proach
works
for
a
function
of
two
variables,
equivalent
to
the
intensive
form
of
U
in
a
one-component
system.
Example
6.2
Demonstration
of
Eq.
6.22
Demonstrate
Eq.
6.22
for
a
function
of
two
variables,
y (0) = f (x1 ,x2 )
.
!
The
second-order
variation
of
f
is:
(0) 2
(0)
(0) 2
2f = 2y (0) = y11
x1 + 2y12
x1x2 + y22
x2
!
The
auxiliary
variables
of
Eq.
6.21
are,
in
this
case:
(1)
z = x1 + y12
x2
! 1
Beegle,
B.
L.,
Modell
M.,
and
Reid,
R.
C.,
AIChE
J.
20:1200-6
(1974).
10/4/12 version
165
6.2 Stability Criteria
z = x2
! 2
Eq.
6.22
can
be
written
explicitly
as:
2
ykk(k1) zk2
!k=1
(0)
(1)
(0)
(1)
(1)
= y11
z12 + y22
z22 = y11
x1 + y12
x2 + y22
x22
(i)
We
now
show
that
this
expression
can
be
reduced
to
Eq.
6.20,
using
the
step-
down
relationships
of
4.5:
(1)
y11
=
1
(0)
y11
(1)
!!!;!!!!y12
=
(0)
y12
(0)
y11
(1)
(0)
!!!;!!!!y22
= y22
( )!
(0)
y12
(0)
y11
Substituting in (i) above,
( )
2
2
(0)
(0)
y12
y
ykk(k1) zk2 = y11(0) x1 + y12(0) x2 + y22(0) y(0) x22 =
k=1
11
11
(y ) x
x +
(0)
(0)
= y11
x12 + 2y12
(0)
12
(0)
y11
2
2
2
(0)
+y22
x22
(y ) x
(0)
12
(0)
y11
2
2
(0)
(0)
(0)
= y11
x12 + 2y12
x2 +y22
x22 = yij(0) xi x j !!!!!!!!!QED
i=1 j=1
Given
that
Eq.
6.20
is
a
quadratic
form,
we
can
now
state
that
the
necessary
and
sufficient
conditions
for
stability
in
a
general
n-component
system
are
that
the
fol-
lowing
n+1
second
derivatives
of
the
basis
function
and
its
Legendre
transforms
are
positive:
y (k1) > 0!!!for!k = 1,2,n +1
! kk
(6.23)
Reordering
the
variables
of
the
basis
function
leads
to
a
different
(but
equiva-
lent)
set
of
stability
conditions.
For
example,
for
a
one-component
system,
the
fol-
lowing
is
a
non-exhaustive
list
of
equivalent
sets
of
stability
conditions:
P
T
(0) T
(1)
y (0) = U(S,V ,N) y11
=
=
> 0!!!;!!!y22
= > 0
(6.24)
S V ,N NCV
V T ,N
10/4/12 version
166
Chapter 6. Equilibrium, Stability, and Fluctuations
T
(0) T
(1)
y (0) = U(S,N ,V ) y11
=
=
> 0!!!;!!!y22
=
>
0
(6.25)
S N ,V NCV
N T ,V
P
T
(0)
(1) T
y (0) = U(V ,S,N) y11
= > 0!!!;!!!y22
= =
> 0
(6.26)
V S ,N
S P ,N NC P
(0)
(1) T
y (0) = U(N ,S,V ) y11
=
>
0!!!;!!!y
=
>
0
S
22
N S ,V
,V
(6.27)
Thus,
thermodynamic
stability
requires
that
all
the
second
derivatives
of
funda-
mental
equations
with
respect
to
a
single
one
of
their
extensive
natural
variables
to
be
positive,
since
an
ordering
of
variables
can
be
generated
to
render
them
in
the
(k1)
form ykk
.
Second
derivatives
of
transforms
with
respect
to
intensive
variables
!
for
not
give
valid
stability
conditions.
For
example,
the
second
derivative
of
A
with
respect
to
the
intensive
variable
T,
2 A
S
2 =
T V ,N
T V ,N
!
does
not
give
a
valid
stability
condition.
This
derivative
is
negative
according
to
Eq.
6.25.
Only
extensive
variables
provide
stability
conditions
because
they
are
the
only
ones
that
obey
conservation
laws
such
as
those
given
by
Eq.
6.12.
Thus
far,
we
have
determined
a
set
of
n+1
thermodynamic
derivatives
that
are
required
to
be
positive
for
a
stable
n-component
system.
If
we
know
that
we
are
already
in
a
stable
thermodynamic
region,
we
now
show
that
we
can
anticipate
which
one
of
the
n+1
thermodynamic
stability
conditions
will
be
violated
first
when
approaching
a
limit
of
stability.
We
have
seen
in
4.5
that
there
is
a
step-
down
relationship
linking
derivatives
of
successive
transforms.
Applying
Eq.
4.31,
(k1)
(k2)
we
can
obtain
ykk
in
terms
of
ykk
:
!
!
(k1)
(k2)
ykk
= ykk
(y ) !!!!k > 2
(k2)
k1,k
(k2)
yk1,k1
(6.28)
We have already established that second derivatives of transforms with respect to
a single extensive natural variable are positive in a stable thermodynamic system.
(k2)
(k2)
Thus, we know that both ykk
and yk1,k1
are positive, since variables k and k1
!
(k2)
!
are untransformed (extensive) in the transform of order k2. If yk1,k1
were to
!
approach zero, then the second, negative term in Eq. 6.28 would dominate the sum
Beegle, B. L.; Modell, M.; and Reid, R. C., AIChE J., 20: 1200-6 (1974).
10/4/12 version
167
6.2 Stability Criteria
(k1)
and
make
ykk
itself
to
become
negative.
Thus,
we
conclude
that
derivatives
of
!
higher-order
transforms
approach
zero
ahead
of
derivatives
of
lower-order
trans-
forms,
when
starting
from
a
stable
region.
Thus,
we
can
establish
that
a
single
sta-
bility
condition
(the
second
derivative
of
the
highest-order
transform)
is
first
vio-
lated
on
the
limit
of
stability
for
a
system
of
n
components:
(n)
yn+1,n+1
> 0
We
will
first
use
this
concept
to
identify
a
pecking
order
among
stability
con-
ditions
for
a
one-component
system.
A
condition
corresponding
to
a
first-order
transform,
such
as
2 A
P
P
(1)
y (0) = U(S,V ,N) y22
= 2 > 0 > 0 < 0
(6.29)
V T ,N
V T
V T ,N
!
is violated ahead of conditions corresponding to the basis function, such as
2 U
T
T
(0)
y (0) = U(S,V ,N) y11
= 2 > 0
>0
> 0
CV
S V ,N
S V ,N
!
(6.30)
Eq.
6.29
is
called
the
condition
of
mechanical
stability
and
implies
that
pres-
sure
decreases
as
volume
increases
at
constant
temperature,
or
equivalently
that
the
isothermal
compressibility
T
>
0.
Eq.
6.30
is
called
the
thermal
stability
condi-
tion.
The
constant-volume
heat
capacity
remains
finite
at
the
limit
of
stability.
An-
other
key
stability
condition
for
a
one-component
system
is
obtained
from
the
en-
thalpy
first-order
transform:
2 H
T
T
(1)
y (0) = U(V ,S,N) y22
= 2 > 0 > 0
> 0
(6.31)
CP
S P ,N
S P ,N
!
As
will
be
shown
in
Example
6.3,
the
two
conditions
represented
by
Eqs.
6.29
and
6.31
are
equivalent;
they
get
violated
simultaneously
when
a
system
reaches
a
limit
of
stability.
For
binary
systems,
the
key
condition
that
is
first
violated
at
a
limit
of
stability
(2)
is
the
one
resulting
from
y33
,
e.g.:
!
2 G
(2)
y (0) = U(S ,V ,N1 ,N2 ) y33
=
> 0 1
> 0
(6.32)
2
N
N1 T ,P ,N
1 T ,P ,N
2
2
!
The
condition
of
Eq.
6.32
is
commonly
used
to
determine
whether
a
binary
system
separates
into
two
immiscible
phases
at
a
given
temperature
and
pressure,
as
dis-
10/4/12 version
key
stability
condition
for
n-component
system
168
Chapter 6. Equilibrium, Stability, and Fluctuations
cussed
in
Chapter
9.
Other
equivalent
key
stability
conditions
for
binary
systems
include:
(2)
y (0) = U(S,V ,N2 ,N1 ) y33
= 2
> 0
N2 T ,P ,N
1
!
(2) T
y (0) = U(N1 ,N2 ,S,V ) y33
=
> 0
S , ,V
1 2
!
P
(2)
y (0) = U(N1 ,N2 ,V ,S) y33
=
>0
V , ,S
1 2
!
Example 6.3 Equivalence of stability criteria
Prove that the key stability conditions for a one-component system given by Eqs.
6.29 and 6.31 are equivalent, by expressing these two derivatives in terms of each
other.
The two relevant derivatives are:
2 A
2 H
P
T
1 P
1 T
T
and
=
=
=
= =
2
2
N V T
V T ,N
S P ,N S P ,N N S P NC P
V T ,N
!
!
Both
are
second
derivatives
of
a
fundamental
equation
with
respect
to
one
of
its
natural
variables.
To
express
them
in
terms
of
each
other,
we
need
to
introduce
another
variable
using
the
chain
rule,
Eq.
5.11:
P
P S
=
V T S T V T
!
(i)
The
first
term
of
the
right-hand
side
of
expression
(i)
can
be
obtained
from
the
XYZ-1
rule
as:
P S T
P
T P
S T P = 1
S = S T
T P S
T
P S
inversion!rule
!
Substituting
(ii)
into
(i):
P
T P S
P
T
V = S T V
T
P S
T
V T S P
!
!
10/4/12 version
(ii)
169
6.2 Stability Criteria
The
two
derivatives
of
interest
are
proportional
to
each
other;
when
one
of
them
is
zero
the
other
one
is
zero
as
well.
Example
6.4
Heating
on
adiabatic
expansion
In
3.1
it
was
suggested
that,
unlike
ideal
gases,
some
substances
heat
up
when
expanded
adiabatically.
Express
the
relevant
thermodynamic
relationship
in
terms
of
derivatives
of
known
sign
(as
far
as
possible)
and
determine
the
requirements
on
material
properties
for
a
substance
to
heat
up
when
expanded
adiabatically.
The
relevant
thermodynamic
derivative
for
determining
the
adiabatic
change
of
temperature
with
volume
is
(T/V) S
.
We
need
to
express
it
in
terms
of
deriva-
tives
that
have
known
signs
in
stable
thermodynamic
systems.
For
this,
we
use
the
chain
rule
(Eq.
5.11)
and
the
triple-product
rule
(Eq.
5.12)
to
obtain:
(
(
)
)
)(
T / P
S / P T / S
T
S
T
P
=
=
V
S V / P
V / P
S
S
!
The
denominator
of
the
right-hand
side
of
(i),
(i)
(V / P)S
!
is
always
negative,
according
to
Eq.
6.18.
The
derivative
(T / S)P = (T / S)P.N
!
is
always
positive
according
to
Eq.
6.26.
The
other
derivative
can
be
transformed
using
Maxwells
rule
on
G:
dG = SdT +VdP (S / P)T = (V / T )P = V P
!
Since
the
coefficient
of
thermal
expansion
P
can
be
either
positive
or
negative,
the
derivative
in
(i)
can
have
either
sign.
The
more
common
(negative)
sign
occurs
for
substances
that
have
positive
coefficient
of
thermal
expansion
P,
while
the
oppo-
site
is
true
for
substances
with
negative
P
.
Ideal
gases
always
have
positive
coeffi-
cients
of
thermal
expansion.
6.3
Fluctuations
Our
discussion
of
equilibrium
conditions
in
6.1
has
established
that
for
a
given
set
of
external
variables
(or
constraints)
on
a
system,
the
corresponding
Legendre
transformation
is
minimized
or
maximized
at
equilibrium,
depending
on
whether
the
starting
point
was
the
energy
of
the
entropy.
In
the
previous
section,
we
analyzed
stability
from
a
macroscopic
point
of
view,
establishing
sets
of
stability
10/4/12 version
170
Chapter 6. Equilibrium, Stability, and Fluctuations
conditions for thermodynamic systems expressed as second derivatives of
thermodynamic potential functions. At a microscopic level, even for a stable
thermodynamic system, the unconstrained variables of a system undergo
continuous fluctuations around their mean values. These fluctuations will turn out
to be intimately linked to the second derivatives of the corresponding partition
function for the relevant statistical ensemble.
As a first step, let us compute the mean-squared fluctuation of energy in the
canonical (NVT) ensemble. In this ensemble, probabilities of microstates are:
exp U
P =
!!!!!where!!!!!Q (N ,V ,T ) =
exp U
Q
all!microstates!!
!
The
mean-squared
fluctuations
of
energy
are:
(U)2 = (U U)2 = U 2 2UU + U2 = U 2 U2 =
=
PU 2
all!microstates!
PU
all!microstates!
The sums can be related to the first and second derivatives of !Q (N ,V ,T ) :
=
N ,V
all!microstates!!
2 Q
=
2
N ,V
!
U exp U = Q
all!microstates!!
U 2 exp
( U ) =
PU
all!microstates!!
Q
PU 2
all!microstates!!
So
that:
1 2 Q
(U) = 2
Q
N ,V
!
2
1 Q
2 lnQ
U
=
=
2
Q
N ,V
N ,V
N ,V
The
last
equality
results
from
the
fact
that
the
logarithm
of
the
canonical
partition
function
is
the
first
Legendre
transform
of
S /kB .
Now
using
the
definition
of
the
!
heat
capacity
at
constant
volume,
U
(U)2 = kBT 2
= kBT 2NCV
T N ,V
!
(6.33)
This is a remarkable result. We have obtained a relationship between the
thermodynamic parameters of a system and the magnitude of spontaneous fluctu-
ations. In the language of Legendre transformations, we have shown that for
y (1) = lnQ = y (1)(,V ,N) , the mean-squared fluctuations of the energy are equal
!
(1)
to the corresponding second derivative, y11
.
!
10/4/12 version
171
6.3 Fluctuations
It
is
interesting
to
note
that
the
heat
capacity
of
the
system,
CV,
is
an
intensive
variable
(independent
of
the
size
of
the
system),
so
the
absolute
magnitude
of
the
mean-squared
fluctuations
grows
linearly
with
system
size.
However,
more
rele-
vant
is
the
magnitude
of
the
fluctuations
relative
to
the
mean
energy
of
the
system:
(U)2
=
U
kBT 2NCV
U
N
1
=
N
N
(6.34)
23
For a macroscopic system, N = O(10 ), so the relative fluctuations are extremely
small on average: the energy of an ideal gas system at equilibrium with a room-
11
temperature bath is constant to roughly 1 part in 10 . However, simulations are
often performed with very small systems having as few as 100 molecules. The
thermal energy of molecules is of O(kBT), and the heat capacities are of O(kB), so
typical relative fluctuations in energy are of the order of 10%.
We can easily generalize the fluctuation relationships to other ensembles. For
example, in the grand canonical (VT) ensemble for a one-component system, fluc-
tuations in the number of molecules are obtained from the second derivative of the
partition function of the ensemble (see 5.5):
y (0) = ln(U ,N ,V )!!;!!y (2) = ln(,,V )
2 ln
N
N
(2)
(N)2 = y22
=
=
=
k
T
B
2
() ,V () ,V
T ,V
!
(6.35)
Similarly, fluctuations in the total volume of a system under NPT conditions
can be obtained as follows:
y (0) = ln(U ,V ,N)!!;!!y (2) = ln(,P,N)
2 ln
V
V
(2)
(V )2 = y22
=
=
= kBT
2
(P) ,N
P T ,N
(P) ,N
!
(6.36)
It is interesting to note that there is a direct connection between microscopic
fluctuations and stability conditions. Fluctuations of the energy in the canonical
ensemble were found to be proportional to the heat capacity at constant volume,
which is positive for stable thermodynamic systems. Fluctuations of the number of
molecules in the grand canonical (VT) ensemble, and of the volume in the NPT
ensemble, are also proportional to derivatives that are positive for stable systems.
Moreover, these latter derivatives diverge at a limit of stability for a one-
component system. Thus, the microscopic interpretation of the loss of stability is
the development of unbounded fluctuations within a system. Conversely, one can
interpret the macroscopic stability conditions as having a positive sign because
they represent mean-squared fluctuations, quantities that are necessarily positive
in sign.
10/4/12 version
172
Chapter 6. Equilibrium, Stability, and Fluctuations
Example 6.5 Fluctuations involving uncorrelated particles**
Compute the mean-squared fluctuations in the number of particles for an open
system at a very low density ! = N /V , for which the particles can be considered
to be uncorrelated. Obtain the equation of state for such a system.
We divide an open system of total volume V, containing <N> particles on average,
into !m N subcells, so that each one of them unlikely to contain more than one
particles at any given time. We denote by ni the cell occupancy of subcell i, with
!
m
ni = 0!!or!!1,!!!and!!! n i = N
i=1
!
The
mean-squared
fluctuation
of
the
total
number
of
particles
in
V
is:
(N)2 =
!
( N N)
= N 2 N2 = ni n j ni n j
By
interchanging
the
averages
and
the
sums
(since
average
of
a
sum
is
always
equal
to
the
sum
of
averages),
m m
< (N)2 >= < ni n j > < ni >< n j >
i=1 j=1
!
Since two different cells are completely independent, we must have
< n n >=< ni >< n j > for i j and the only terms that do not cancel out in the double
! i j
sum are ones with i = j :
m
< (N)2 >= (< ni2 > < ni >2 )
i=1
!
Since ni = 0 or 1, ni2 = ni , so that
m
(N)2 = ni ni 2 = m ni ni 2 = m n1 (1 n1 ) m n1 = N
i=1
From
this
equation
and
the
general
expression
for
fluctuations
in
the
grand
canon-
ical
ensemble,
we
can
obtain
information
on
the
thermodynamic
functions
of
the
system:
(N)2 =
1 N
= N
,V
**
Following
3.6
in
Chandler,
D.,
Introduction
to
Modern
Statistical
Mechanics,
Oxford
Univ.
Press, 1987.
10/4/12 version
173
6.3 Fluctuations
Dividing by V and taking the reciprocal, with ! = N /V
1
= = = constant + ln
,V
This
is
the
expression
in
classical
thermodynamics
for
the
chemical
potential
for
an
ideal
gas.
From
!d = dG = SdT +VdP ,
with
=
1/V,
=
1/kBT
P
1 P
1
= = P = + constant
The
integration
constant
for
P
is
0
since
at
=
0,
P
=
0.
Eq.
[33]
is
then
just
the
ide-
al
gas
law:
P = PV = NkBT , (where N is the number of molecules, not moles)
!
The purpose of this example is to show that very simple microscopic assumptions
(independence of all particles) result in useful, experimentally valid expressions
for the macroscopic thermodynamic behavior.
6.4 Maxwell Construction, Binodals and Spinodals
When pressure is plotted against the specific volume, experimentally deter-
mined isotherms for many systems follow paths similar to those shown in Fig. 6.7,
with a horizontal portion marking a phase transition, defined as a sharp disconti-
nuity in a thermodynamic proper-
ty. For this specific example, the
low-volume, high-pressure phase
on the left branch of the isotherm
is a liquid, and the high-volume,
low-pressure phase on the right
branch a vapor, so the horizontal
portion corresponds to vaporiza-
tion of the liquid or condensation
of the vapor. Provided that care is
taken to eliminate sources of nu-
cleation, the liquid and vapor iso-
therms can be measured into the
Figure 6.7 Schematic of an experimentally de-
metastable regions, indicated by
termined isotherm for a system with a liquid-
the dashed lines in Fig. 6.7. The vapor phase transition. Dashed lines are metasta-
ble extensions of the liquid and vapor branches.
saturated liquid and vapor phases
are at the two ends of the horizon-
10/4/12 version
174
Chapter 6. Equilibrium, Stability, and Fluctuations
tal line segment. Thermodynamic
theories and engineering equa-
tions of state of the type discussed
in the next chapter, on the other
hand, produce isotherms that con-
nect the liquid and vapor branches
by a smooth curve, as shown in
Fig. 6.7. This curve is called a van
der Waals loop. The points marked
1 and 2 in this figure are limits of
stability, since for these points,
Figure
6.7
Schematic
of
a
model
or
equation-of-
state
isotherm
for
a
system
with
a
phase
transi-
tion.
The
stability
condition
is
violated
along
the
dotted
line.
P
V = 0
T
!
(6.37)
The
van
der
Waals
loop
violates
the
mechanical
stability
condition,
Eq.
6.24,
along
the
dotted
line
between
points
1
and
2.
We
know
that
real
systems
cannot
exist
at
such
states,
but
the
presence
of
a
continuous
path
between
phases
in
equation-of-
state
isotherms
provides
a
mechanism
for
satisfying
the
conditions
of
equilibrium.
In
other
words,
we
can
use
the
model
isotherm
of
Fig.
6.7
to
determine
the
points
along
it
that
satisfy
equality
of
chemical
potentials,
L
=
V
.
Starting
from
the
fun-
damental
equation
for
the
Gibbs
free
energy,
for
any
two
points
along
an
isotherm,
(6.38)
P = V = V dP
T
!
In order to see what this relationship im-
plies, let us flip the axes of Fig. 6.7 to obtain Fig.
6.8. We now draw a vertical line that leaves
equal areas to the left and right of it, as shown in
the figure. For equal areas, the integral of !VdP
from point A to point B is zero, so Eq. 6.38 sug-
gests that the chemical potentials are equal at A
and B. These two points also have equal pres-
sures (being on the same vertical line) and equal
temperatures (being on the same isotherm).
They satisfy the conditions of phase coexistence
(Eq. 6.5) and the coexisting phases can now be
identified as A L (saturated liquid) and B V
10/4/12 version
Figure 6.8 Maxwell construction.
6.4 Maxwell Construction, Binodals and Spinodals
175
(saturated vapor). This procedure is known as Maxwell construction, named after
the Scottish physicist James Clerk Maxwell who first proposed it. Line segments 1A
and 2B (dashed) are metastable states, satisfying the stability condition but be-
yond the points of stable equilibrium. They correspond to the dashed extensions of
the stable liquid and vapor branches seen in experimental systems (Fig. 6.7, p.
173).
The densities and other properties of coexisting phases depend on tempera-
ture. For the vapor-liquid transition, the two phases become increasingly more
similar as temperature is raised. At a very special location in thermodynamic co-
ordinate space called the critical point, the properties of the two phases at coexist-
ence become identical. Such critical points are seen not only for vapor-liquid tran-
sitions, but for liquid-liquid equilibria as well. Many other physical phenomena can
be described as phase transitions ending at critical points for example, the Curie
point, a temperature above which magnets lose their ability to retain a permanent
magnetization, is also a critical point.
A schematic phase diagram showing two subcritical isotherms as well as the
critical isotherm is presented in Fig. 6.9. As previously, stable segments of the iso-
therms are shown as continuous lines, metastable segments shown as dashed
lines, and unstable segments as dotted lines. The coexistence points are obtained
from Maxwell constructions at each temperature. The locus of coexistence points
defines the vapor-liquid coexistence envelope, which is also known as the binodal
curve. The locus of stability limits (points along which the condition of zero deriva-
tive of the isotherms, Eq. 6.37, holds) define the spinodal curve and the region
within this curve is known as the spinodal region. The spinodal curve is entirely
within the two-phase equilibrium (binodal) curve; the two meet at the critical
point. Within the spinodal region (horizontally hatched area), the system is unsta-
ble with respect to phase separation; real systems rapidly form two phases, in a
Figure
6.9
Spinodal
(horizontally
hatched)
and
metastable
(diagonally
hatched)
regions
with-
in
the
coexistence
(binodal)
curve.
10/4/12 version
176
Chapter 6. Equilibrium, Stability, and Fluctuations
process
known
as
spinodal
decomposition.
The
two
diagonally
hatched
areas
be-
tween
the
spinodal
and
binodal
curves
are
metastable
regions
for
the
liquid
(left,
low
V
)
and
gas
(right,
high
V
).
In
these
regions,
formation
of
a
two-phase
system
is
thermodynamically
favored
but
requires
nucleation
and
growth
of
the
new
phase.
As
discussed
in
6.2,
the
vanishing
of
the
first
derivative
of
the
pressure
with
respect
to
volume
defines
a
limit
of
stability
because
it
corresponds
to
a
second
derivative
of
the
Helmholtz
free
energy
A.
However,
at
the
critical
point,
the
se-
cond
derivative
of
the
pressure
with
respect
to
volume
also
vanishes
this
is
be-
cause
the
critical
point
represents
the
merging
of
a
maximum
and
a
minimum
of
the
P
versus
V
isothermal
curves.
At
a
minimum
(left
side
of
the
spinodal
in
Fig.
6.9)
the
second
derivative
is
positive
and
at
a
maximum
(right
side
of
the
spinodal)
the
second
derivative
is
negative.
Since
the
isotherm
itself
is
continuous
and
has
no
discontinuities
in
its
derivatives,
this
can
only
happen
if
the
second
derivative
is
zero
at
the
critical
point.
Stability
requires
that
the
next
derivative
(third
deriva-
tive
of
pressure
with
respect
to
volume)
is
positive,
a
condition
satisfied
at
the
crit-
ical
point.
The
criticality
conditions
are
then:
P P 2
V = 2 = 0
T V T
!
(6.39)
We have shown in Example 6.3 that the two stability conditions:
P
T
> 0
and
< 0
CP
V T
!
are equivalent. Therefore, at any critical point,
P
T
V = 0 C = 0 CP
P
T
(6.40)
Many
other
physical
quantities
diverge
at
critical
points.
For
example,
density
fluc-
tuations
in
a
normal
liquid
or
gas
are
correlated
only
over
length
scales
compa-
rable
to
the
size
of
molecules
and
thus
much
shorter
than
the
wavelength
of
light.
Near
critical
points,
the
correlation
length
for
density
fluctuations
increases
rapid-
ly
and
becomes
much
bigger
than
molecular
size.
This
gives
rise
to
a
characteristic
milky
appearance
of
fluids
near
criticality,
known
as
critical
opalescence.
6.5 Gibbs Phase Rule
The equilibrium criteria of Eq. 6.5 are that pressure, temperature and chemical
potential for each component should be uniform throughout a system at equilibri-
um. We know from everyday experience that multiple phases of different densities
10/4/12 version
177
6.5 Gibbs Phase Rule
can
exist
at
different
conditions
for
example,
water
is
found
in
nature
as
solid
ice,
liquid
water
and
vapor.
In
6.3
we
suggested
that
two
phases
could
coexist
for
a
range
of
temperatures
at
a
specific
pressure
for
one-component
systems.
An
inter-
esting
question
with
important
practical
implications
is
whether
the
conditions
of
equilibrium
impose
restrictions
on
the
number
of
possible
phases
that
can
simul-
taneously
be
present
in
a
system
at
equilibrium.
The
answer
turns
out
to
depend
on
the
number
of
independent
chemical
species
(components)
in
a
system,
in
a
relationship
first
proposed
by
J.
Willard
Gibbs
in
1870.
We
have
seen
in
Chapter
5
that
n+2
thermodynamic
variables
are
required
to
fully
specify
the
state
of
a
thermodynamic
system.
These
could
be
selected
as
(T,P,N 1 ,N 2 ,,N n ).
Dividing
the
molar
amounts
by
the
total
number
of
moles
in
the
system,
N
=
N 1 +N 2 ++N n ,
we
can
express
thermodynamic
properties
in
terms
of
(T,P,x 1 ,x 2 ,,x n1,N).
However,
if
we
restrict
our
attention
to
just
intensive
properties,
we
can
eliminate
N
from
the
variable
set
and
express
these
properties
in
terms
of
n+1
intensive
variables,
(T,P,x 1 ,x 2 ,,x n1).
Now
consider
a
system
with
phases
coexisting
at
equilibrium.
We
denote
by
x i , j
the
mole
fraction
of
component
i
in
phase
j
and
by
i , j
the
chemical
potential
of
component
i
in
phase
j.
The
conditions
imposed
by
equilibrium
on
the
chemical
potentials
at
coexistence,
Eq.
6.6,
can
now
be
written
as:
1,1 = 1,2 = = 1,
2,1 = 2,2 = = 2,
(6.41)
= n,2 = = n,
! n,1
There
are
!n( 1)
independent
equations
in
this
set
of
relationships.
Each
of
the
chemical
potentials
in
Eqs.
6.41
can
be
written
as
a
function
of
the
tempera-
ture,
pressure
and
composition
variables
in
each
phase:
1,1 = 1,1 (T ,P,x1,1 ,x2,1 ,x n1,1 )
2,1 = 2,1 (T ,P,x1,1 ,x2,1 ,x n1,1 )
n,1 = n,1 (T ,P,x1,1 ,x2,1 ,x n1,1 )
1,2 = 1,2(T ,P,x1,2 ,x2,2 ,x n1,2 )
2,2 = 2,2(T ,P,x1,2 ,x2,2 ,x n1,2 )
= n, (T ,P,x1, ,x2, ,x n1, )
! n,
10/4/12 version
(6.42)
178
Chapter 6. Equilibrium, Stability, and Fluctuations
Thus,
there
are
!2+(n 1)
intensive
variables
describing
the
chemical
po-
tentials,
the
common
T
and
P
as
well
as
(n1)
mole
fractions
for
each
phase.
But
these
are
not
all
independent,
as
they
must
satisfy
the
!n ( 1)
relationships
of
Eq.
6.42.
The
number
of
independent
intensive
variables,
which
is
defined
as
the
number
of
degrees
of
freedom,
F ,
of
the
system
is:
F = !{number!of!total!variables}! !{number!of!constraints}!
F = 2+(n1) n( 1) = 2+ n n + n
!F = n + 2
Gibbs
(6.43)
phase
rule
Phase
diagrams
of
pure
components
often
contain
multiple
solid
phases
(pol-
ymorphs),
as
illustrated
in
Fig.
6.10,
which
shows
a
partial
high-pressure
phase
Figure
6.10
Partial
phase
diagram
of
water
at
high
pressures.
Triple
points
are
shown
as
circles.
Data
are
from
Revised
Release
on
the
Pressure
along
the
Melting
a nd
Sublimation
Curves
of
Ordinary
Water
Substance,
International
Association
of
Properties
of
Water
and
Steam,
(2008)
and
L.
Mercury
et
al.,
Thermodynamics
of
ice
polymorphs
and
`ice-like'
water
in
hydrates
and
hydroxides,,
Appl.
Geochem.
16,
161-181
(2001).
10/4/12 version
179
6.5 Gibbs Phase Rule
diagram
of
water,
including
multiple
solid
polymorphs.
Ice
formed
from
liquid
wa-
ter
at
near-atmospheric
conditions
is
hexagonal
ice
Ih.
The
phase
rule
does
not
impose
a
limit
on
the
number
of
possible
phases
pre-
sent
in
different
locations
of
the
phase
diagram,
it
only
restricts
how
many
can
co-
exist
at
any
given
temperature
and
pressure.
As
suggested
by
the
phase
rule,
areas
in
this
diagram
contain
only
a
single
phase
(F
=
2,
two
degrees
of
freedom,
corre-
sponding
to
two-dimensional
geometric
objects),
while
lines
(F
=
1)
correspond
to
coexistence
of
two
phases.
Three
phases
coexist
at
triple
points
(F
=
0).
A
one-
component
system
cannot
exist
as
four
separate
phases
at
any
point
in
the
phase
diagram,
as
this
corresponds
to
negative
(unfeasible)
degrees
of
freedom.
Another
case
of
interest
is
binary
systems.
Intensive
variables
of
interest
for
binary
systems
are
the
temperature,
pressure
and
composition
(mole
fraction
of
one
component).
Single
phase,
two-component
systems
have
F
=
3,
three
degrees
of
freedom,
corresponding
to
volumes
in
the
three-dimensional
space
of
the
inten-
sive
variables.
Application
of
the
phase
rule
to
two-phase
systems
gives
F
=
2,
two
degrees
of
freedom,
so
that
at
a
given
temperature
and
pressure
a
range
of
compo-
sitions
can
exist.
For
three-phase
systems,
there
is
a
line
of
triple
points
(F
=
1),
along
which
temperature,
pressure
and
composition
varies.
The
conditions
of
criticality
(Eq.
6.39)
impose
two
additional
constraints
on
an
otherwise
one-phase
system,
thus
using
up
two
degrees
of
freedom.
Thus,
for
a
one-component
system
the
number
of
degrees
of
freedom
at
criticality
is
zero,
so
we
are
justified
to
talk
about
a
critical
point.
For
two-component
systems,
there
is
a
critical
line
along
which
the
critical
composition,
pressure
and
temperature
vary.
10/4/12 version
Das könnte Ihnen auch gefallen
- Chapter 4 Bearings PDFDokument50 SeitenChapter 4 Bearings PDFAnonymous 7aN0oYUm7vNoch keine Bewertungen
- Isa S75 01 PDFDokument52 SeitenIsa S75 01 PDFAnton HidayatNoch keine Bewertungen
- Optimal Operation of A Semi-Batch Reactive Distillation Column (2000)Dokument7 SeitenOptimal Operation of A Semi-Batch Reactive Distillation Column (2000)GodofredoNoch keine Bewertungen
- Bettering Operation of Robots by Learning, Arimoto Et AlDokument18 SeitenBettering Operation of Robots by Learning, Arimoto Et AlPeps Peps PepsNoch keine Bewertungen
- Fluid MechanicsDokument40 SeitenFluid MechanicsShahinur AlamNoch keine Bewertungen
- Physics Formula-Book JeeDokument115 SeitenPhysics Formula-Book JeePritish Jaiswal82% (17)
- GATEFLIX Chemical Reaction EngineeringDokument129 SeitenGATEFLIX Chemical Reaction EngineeringVijaykumar JatothNoch keine Bewertungen
- Analysis and Design of Air-Jet Pumps For Pneumatic TransportationDokument302 SeitenAnalysis and Design of Air-Jet Pumps For Pneumatic TransportationMusheer BashaNoch keine Bewertungen
- CHE 312 Lecture 2Dokument17 SeitenCHE 312 Lecture 2Muhammad Hamza EjazNoch keine Bewertungen
- Chapter 2Dokument51 SeitenChapter 2yungP100% (2)
- Introduction To DimensionalAnalaysis (Daw)Dokument26 SeitenIntroduction To DimensionalAnalaysis (Daw)tahadostmushtaqNoch keine Bewertungen
- Transport Phenomena AssignmentDokument3 SeitenTransport Phenomena AssignmentKushal KayalNoch keine Bewertungen
- W1 Energy and Energy BalanceDokument32 SeitenW1 Energy and Energy BalanceFatinnnnnnNoch keine Bewertungen
- 2 πN 60 = 2 π (1200 rev) 60 = 40 π 18 μDokument3 Seiten2 πN 60 = 2 π (1200 rev) 60 = 40 π 18 μNUR ASYIQIN BINTI AZAHARNoch keine Bewertungen
- Soal SoalDokument8 SeitenSoal SoalJakaAbuNoch keine Bewertungen
- Gases and Vacua: Handbook of Vacuum PhysicsVon EverandGases and Vacua: Handbook of Vacuum PhysicsA. H. BeckNoch keine Bewertungen
- CRE 1-3 Unit (2016-2017) PDFDokument56 SeitenCRE 1-3 Unit (2016-2017) PDFgouthamNoch keine Bewertungen
- Process Dynamics and Control 3rd Edition Chapter 12 Problem 3E SolutionDokument8 SeitenProcess Dynamics and Control 3rd Edition Chapter 12 Problem 3E Solutioneduardo acunia0% (1)
- Pinch Technology by ShubhamDokument29 SeitenPinch Technology by Shubhamapi-381283580% (5)
- Practical Chemical Thermodynamics for GeoscientistsVon EverandPractical Chemical Thermodynamics for GeoscientistsNoch keine Bewertungen
- 1-1 Unit (2016-2017)Dokument113 Seiten1-1 Unit (2016-2017)goutham100% (1)
- Modeling in Transport Phenomena: A Conceptual ApproachVon EverandModeling in Transport Phenomena: A Conceptual ApproachBewertung: 3 von 5 Sternen3/5 (2)
- Principles 3 PDFDokument26 SeitenPrinciples 3 PDFHasan AkhuamariNoch keine Bewertungen
- (4.1) Laminar Premixed FlameDokument31 Seiten(4.1) Laminar Premixed Flameمصطفى العباديNoch keine Bewertungen
- Scale Up ArticleDokument8 SeitenScale Up ArticleAndrek QueckNoch keine Bewertungen
- Solution ThermoDokument33 SeitenSolution ThermoNur Aqilah100% (1)
- Measurement of Gas Flow Iso 9300Dokument11 SeitenMeasurement of Gas Flow Iso 9300sojin nath vadakkayilNoch keine Bewertungen
- H W5Dokument1 SeiteH W5Bahadır KayaNoch keine Bewertungen
- 7 Fluid Dynamics Tutorial SolutionDokument25 Seiten7 Fluid Dynamics Tutorial SolutionAldhaAhmadyaningratNoch keine Bewertungen
- Applied Chemical Engineering CalculationsDokument7 SeitenApplied Chemical Engineering Calculationsmbolantenaina100% (1)
- A Mixture Containing Equimolar Amounts of BenzeneDokument1 SeiteA Mixture Containing Equimolar Amounts of BenzeneFranciskus Peri0% (1)
- Che 511 HW Set #5 Due Nov 12: Additional InformationDokument2 SeitenChe 511 HW Set #5 Due Nov 12: Additional InformationAshish KotwalNoch keine Bewertungen
- H.W 7Dokument9 SeitenH.W 7Mirvat ShamseddineNoch keine Bewertungen
- The Modeling of Chemical Reactors Chapter 8Dokument44 SeitenThe Modeling of Chemical Reactors Chapter 8Manoaki100% (1)
- Tutorial 1Dokument2 SeitenTutorial 1eddy50% (2)
- Professional Reference Shelf: Sothermal Eactor EsignDokument5 SeitenProfessional Reference Shelf: Sothermal Eactor EsignSourodip GhoshdastidarNoch keine Bewertungen
- Thermodynamic Properties, Equations of State, Methods Used To Describe and Predict Phase EquilibriaDokument20 SeitenThermodynamic Properties, Equations of State, Methods Used To Describe and Predict Phase EquilibriaanisfathimaNoch keine Bewertungen
- Process Dynamics and Control - Ch05Dokument20 SeitenProcess Dynamics and Control - Ch05etayhailuNoch keine Bewertungen
- ChBE3210 Spring2007 Exam2Dokument6 SeitenChBE3210 Spring2007 Exam2Abishek KasturiNoch keine Bewertungen
- Contoh Soal CreDokument11 SeitenContoh Soal CreMuhammad Irfan SalahuddinNoch keine Bewertungen
- Analogies of Transport PropertiesDokument3 SeitenAnalogies of Transport PropertiesAB DevilierNoch keine Bewertungen
- Thermo FinalDokument66 SeitenThermo Finalsossydj75% (4)
- Enthalpy PDFDokument3 SeitenEnthalpy PDFAnonymous NxpnI6jCNoch keine Bewertungen
- Prob 3 CH 3 PDFDokument2 SeitenProb 3 CH 3 PDFvijendra mauryaNoch keine Bewertungen
- Plug Flow Reactor Equilibrium ConversionDokument24 SeitenPlug Flow Reactor Equilibrium Conversionblackbeauty14100% (1)
- CN2116 HW11 Solution (XJP - 2012) FinalDokument9 SeitenCN2116 HW11 Solution (XJP - 2012) FinalPrimus OngNoch keine Bewertungen
- SI Heat 5e Chap02 LectureDokument84 SeitenSI Heat 5e Chap02 Lecturepremnath chakriNoch keine Bewertungen
- Poiseuille Flow: An Internet Book On Fluid DynamicsDokument4 SeitenPoiseuille Flow: An Internet Book On Fluid DynamicsFelix EzomoNoch keine Bewertungen
- Dynamics of ThermometerDokument12 SeitenDynamics of ThermometerSaumya Agrawal100% (1)
- Drying of Process MaterialDokument30 SeitenDrying of Process MaterialsehagendutNoch keine Bewertungen
- Answer For Tutorial 2Dokument9 SeitenAnswer For Tutorial 2Mukul ParasharNoch keine Bewertungen
- Tutorial 1 SolutionDokument6 SeitenTutorial 1 SolutionMihir Kumar MechNoch keine Bewertungen
- Stoichiometry For Systems Involving Recycles PDFDokument7 SeitenStoichiometry For Systems Involving Recycles PDFGlory Usoro100% (1)
- Simulink Development of Single Effect Evaporator Using MatlabDokument12 SeitenSimulink Development of Single Effect Evaporator Using MatlabRhea MambaNoch keine Bewertungen
- 4.material and Energy Balance..Dokument16 Seiten4.material and Energy Balance..cvkalyanNoch keine Bewertungen
- Heat Conduction From A Sphere To A Stagnant FluidDokument3 SeitenHeat Conduction From A Sphere To A Stagnant FluidMitesh WaghelaNoch keine Bewertungen
- Thus, This Term Actually Means A in A Constant-Volume System The Measure of Reaction Rate of Component I BecomesDokument23 SeitenThus, This Term Actually Means A in A Constant-Volume System The Measure of Reaction Rate of Component I Becomesalice Annabelle100% (1)
- Catalyst Characterization - W6Dokument33 SeitenCatalyst Characterization - W6Safitri WulansariNoch keine Bewertungen
- Interphase Mass TransferDokument20 SeitenInterphase Mass TransferDozdi0% (1)
- Gate 2006 PDFDokument21 SeitenGate 2006 PDFVammsy Manikanta SaiNoch keine Bewertungen
- Exam Solution Chemical ProcessesDokument9 SeitenExam Solution Chemical ProcessesInez Karina TanuNoch keine Bewertungen
- (P01, C01, C02, C2, C3) : Confidential EH/JUN 2014/CHE584/594Dokument11 Seiten(P01, C01, C02, C2, C3) : Confidential EH/JUN 2014/CHE584/594Addison JuttieNoch keine Bewertungen
- Assignment Thermal UiTMDokument29 SeitenAssignment Thermal UiTMiwe1234Noch keine Bewertungen
- Dynamic Programming in Chemical Engineering and Process Control by Sanford M RobertsVon EverandDynamic Programming in Chemical Engineering and Process Control by Sanford M RobertsNoch keine Bewertungen
- SLecture 7Dokument6 SeitenSLecture 7Singh AnujNoch keine Bewertungen
- Schedule Pr152Dokument27 SeitenSchedule Pr152Singh AnujNoch keine Bewertungen
- Nuclear Physics.: PH 424 (HI 1 6 9Dokument1 SeiteNuclear Physics.: PH 424 (HI 1 6 9Singh AnujNoch keine Bewertungen
- Nucl Phys ProblemsDokument6 SeitenNucl Phys ProblemsSingh AnujNoch keine Bewertungen
- Pay Structure of EmployeesDokument16 SeitenPay Structure of EmployeesSingh AnujNoch keine Bewertungen
- JMP2015 Rejected ApplicationsDokument2 SeitenJMP2015 Rejected ApplicationsSingh AnujNoch keine Bewertungen
- Gymnasiums, Iit Kanpur Management, Conduct Rules and Guidelines I. ManagementDokument2 SeitenGymnasiums, Iit Kanpur Management, Conduct Rules and Guidelines I. ManagementSingh AnujNoch keine Bewertungen
- III Be Biotech MSR Mo2012 2Dokument4 SeitenIII Be Biotech MSR Mo2012 2Singh AnujNoch keine Bewertungen
- B-Series Propeller PDFDokument63 SeitenB-Series Propeller PDFCakra Wijaya Kusuma RahadiNoch keine Bewertungen
- Civil Engineering: Detailed Solutions ofDokument58 SeitenCivil Engineering: Detailed Solutions ofVivekChaudharyNoch keine Bewertungen
- V-Notch Weir (Dimensional Analysis) Briefing SheetDokument18 SeitenV-Notch Weir (Dimensional Analysis) Briefing SheetDenys Ilyushenko100% (2)
- Porosity Modeling of Brush SealsDokument7 SeitenPorosity Modeling of Brush SealsНиколайNoch keine Bewertungen
- Studying The Flight of A Paper Airplane Using Manual Simulation and Dimensional AnalysisDokument12 SeitenStudying The Flight of A Paper Airplane Using Manual Simulation and Dimensional Analysisarya ladhaniNoch keine Bewertungen
- 10 Type CurvesDokument18 Seiten10 Type CurvesJun Hao HengNoch keine Bewertungen
- A Porous Media Theory For Characterization of Membrane Blood Oxygenation DevicesDokument13 SeitenA Porous Media Theory For Characterization of Membrane Blood Oxygenation DevicesErkanNoch keine Bewertungen
- Agitation Wk9i - CHE503Dokument19 SeitenAgitation Wk9i - CHE503Abdur Rashid0% (1)
- Dimensional AnalysisDokument15 SeitenDimensional AnalysisJimmyCenturionNoch keine Bewertungen
- Physical World, Unit - DimensionsDokument8 SeitenPhysical World, Unit - DimensionsSarthak ShingareNoch keine Bewertungen
- About: YakeenDokument40 SeitenAbout: YakeenHkkNoch keine Bewertungen
- Ch-2-Dimensional AnalysisDokument15 SeitenCh-2-Dimensional AnalysisRefisa JiruNoch keine Bewertungen
- Non Dimensional NumbersDokument16 SeitenNon Dimensional NumbersSharath ChandraNoch keine Bewertungen
- Chap 5 PDFDokument10 SeitenChap 5 PDFاحمدعطيهNoch keine Bewertungen
- Synopsis Report 12me92r03 1Dokument12 SeitenSynopsis Report 12me92r03 1api-423757765Noch keine Bewertungen
- Ch7 Dimensional Analysis and SimilarityDokument29 SeitenCh7 Dimensional Analysis and Similaritya u khanNoch keine Bewertungen
- 2 Depositional Environments As Interpreted From Primary Sedimentary and Stratigraphic SequencesDokument158 Seiten2 Depositional Environments As Interpreted From Primary Sedimentary and Stratigraphic SequencesTefy Soñadora AustralNoch keine Bewertungen
- Cont.: Fluid Mechanics (MR 231) Lecture NotesDokument9 SeitenCont.: Fluid Mechanics (MR 231) Lecture NotesAhmedTahaNoch keine Bewertungen
- 03-Unit and Dimension (Question) 2 (55-60)Dokument7 Seiten03-Unit and Dimension (Question) 2 (55-60)rahul singhNoch keine Bewertungen
- Effects of Physical Parameter Range On Dimensionless Variable Sensitivity in Water Flooding ReservoirsDokument7 SeitenEffects of Physical Parameter Range On Dimensionless Variable Sensitivity in Water Flooding ReservoirschirazNoch keine Bewertungen
- CFD Assignment 2D Incompressible FlowDokument25 SeitenCFD Assignment 2D Incompressible FlowPraphulNoch keine Bewertungen