Das könnte Ihnen auch gefallen
- Bioquimica Devlin 4a EdicionDokument621 SeitenBioquimica Devlin 4a EdicionLuisNoch keine Bewertungen
- Señalizacion Celular PDFDokument48 SeitenSeñalizacion Celular PDFOscar Ivan0% (1)
- Bioquimica Stryer 6 Edicion (Librosmedicospdf - Net)Dokument1.137 SeitenBioquimica Stryer 6 Edicion (Librosmedicospdf - Net)Jhordan Castillo DiazNoch keine Bewertungen
- Fisiología de Guyton - CompendioDokument134 SeitenFisiología de Guyton - CompendioAngel Alcivar “AAAS”Noch keine Bewertungen
- ORD SUPLENTE Examen LenguaCastellana LiteraturaIIDokument3 SeitenORD SUPLENTE Examen LenguaCastellana LiteraturaIIRuidong LinNoch keine Bewertungen
- Bioquimica de Ferrier PDFDokument531 SeitenBioquimica de Ferrier PDFCarolina Gómez Mora81% (16)
- Microbiologia Médica - BasualdoDokument1.425 SeitenMicrobiologia Médica - BasualdoEnrique Duarte81% (21)
- Biologia e Introduccion A La Biologia CelularDokument128 SeitenBiologia e Introduccion A La Biologia CelularAngie EstradaNoch keine Bewertungen
- Síntesis Del Grupo HemoDokument9 SeitenSíntesis Del Grupo HemoDario TaimalNoch keine Bewertungen
- LibroDokument60 SeitenLibroIris GonzálezNoch keine Bewertungen
- Apuntes Biologia Celular y TisularDokument152 SeitenApuntes Biologia Celular y TisularFatima Jerez50% (2)
- Pentosas FosfatoDokument39 SeitenPentosas FosfatoKarolNoch keine Bewertungen
- Cortes HistologicosDokument5 SeitenCortes Histologicosveruskha pachecoNoch keine Bewertungen
- Manual de BioquímicaDokument77 SeitenManual de BioquímicaAldair PazNoch keine Bewertungen
- Metabolismo CelularDokument10 SeitenMetabolismo CelularDamaris Basilio DamacenNoch keine Bewertungen
- Costanzo PDFDokument42 SeitenCostanzo PDFKrlos Cruz Avila67% (3)
- Bioquimica Las Bases Moleculares de La Vida McKee 5a EdDokument2 SeitenBioquimica Las Bases Moleculares de La Vida McKee 5a EdRichard Solano33% (24)
- CURTIS Uba Xxi 2021 7ma EdDokument135 SeitenCURTIS Uba Xxi 2021 7ma Edjessica carolineNoch keine Bewertungen
- Preguntas YRreepuestas BioquimicaDokument476 SeitenPreguntas YRreepuestas Bioquimicafelix100% (1)
- Manual de Laboratorio de FisiologiaDokument285 SeitenManual de Laboratorio de FisiologiaAbel Fores100% (2)
- Bioquímica: Conceptos Esenciales. 3 EdiciónDokument2 SeitenBioquímica: Conceptos Esenciales. 3 Ediciónsharon perez0% (1)
- Bioquimica Conceptos Esenciales FeduchiDokument383 SeitenBioquimica Conceptos Esenciales FeduchiTopacio Manrique100% (23)
- Caso Clinico - SeñalizacionDokument1 SeiteCaso Clinico - SeñalizacionEdisonAlonzoPazNoch keine Bewertungen
- Anatomia Con Orientacion Clinica de Moore 7ma EdicionDokument10 SeitenAnatomia Con Orientacion Clinica de Moore 7ma Edicionclagadipa25% (8)
- Biología Molecular de La Célula, 5 Edición. Alberts (Español)Dokument10 SeitenBiología Molecular de La Célula, 5 Edición. Alberts (Español)Diego Sebastián C. Olivares0% (1)
- Quimica de RigalliDokument213 SeitenQuimica de RigalliAdrian BrusaNoch keine Bewertungen
- Histologia Basica Texto y Atlas Junqueira CarneiroDokument557 SeitenHistologia Basica Texto y Atlas Junqueira CarneiroDanna Alducin0% (1)
- Bioquimica Humana - Texto y AtlasDokument273 SeitenBioquimica Humana - Texto y AtlasAmbar Gabriela100% (1)
- Bioquimica EnzimasDokument12 SeitenBioquimica EnzimasIlianaMadrigalNoch keine Bewertungen
- LIR. Memorama - BioquímicaDokument396 SeitenLIR. Memorama - BioquímicaNancy Avile100% (10)
- 001 - Acidos y Bases Fuertes-2019 PDFDokument15 Seiten001 - Acidos y Bases Fuertes-2019 PDFlizpaniagua99gmail.com Paniagua100% (1)
- Farmacología: Básica y Clínica 18° Edición VelazquezDokument1.345 SeitenFarmacología: Básica y Clínica 18° Edición VelazquezKarlaCamarilloNoch keine Bewertungen
- Fisiología Humana - Un Enfoque Integrado Silverthron Ed6 PDFDokument1.007 SeitenFisiología Humana - Un Enfoque Integrado Silverthron Ed6 PDFAna Valero97% (38)
- Classificacio EnzimsDokument3 SeitenClassificacio EnzimsjbuedosNoch keine Bewertungen
- Bioquímica y Medicina HarperDokument10 SeitenBioquímica y Medicina HarperAnnete Romero0% (1)
- Ujcm - Facisa - Manual de Prácticas de BioquimicaDokument162 SeitenUjcm - Facisa - Manual de Prácticas de Bioquimicarogelio Martin100% (1)
- LIR Memorama Fisiologia PDFDokument393 SeitenLIR Memorama Fisiologia PDFPepita Sar100% (1)
- Bioquímica HumanaDokument327 SeitenBioquímica HumanaNelson Ivan Yucra94% (35)
- Bioquimica Christopher K Mathews Et All 2013 4ta Ed CompressDokument1.381 SeitenBioquimica Christopher K Mathews Et All 2013 4ta Ed CompressFacundo Exequiel CorreaNoch keine Bewertungen
- Bio Len 7 (1234)Dokument1.234 SeitenBio Len 7 (1234)Isabel VillatoroNoch keine Bewertungen
- Temas Examen de Bioquímica IIDokument9 SeitenTemas Examen de Bioquímica IIManuel Alejandro Iza GutierrezNoch keine Bewertungen
- Bioquimica de Stryer 6 EdiciónDokument1.137 SeitenBioquimica de Stryer 6 EdiciónNicolás Quimbaya70% (74)
- Bioquimica Tercera Edicion Libro de TextDokument6 SeitenBioquimica Tercera Edicion Libro de TextAracely PeñaNoch keine Bewertungen
- Traducción: Transcripción, Traduc Ción Y Replicación CelularDokument1 SeiteTraducción: Transcripción, Traduc Ción Y Replicación CelularNtuys ChNoch keine Bewertungen
- Victoria Sanchez 9C Estructura Del AdnDokument8 SeitenVictoria Sanchez 9C Estructura Del AdnVictoria Sanchez NietoNoch keine Bewertungen
- Organizador Grafico Información GenéticaDokument3 SeitenOrganizador Grafico Información Genéticalquinonez9760Noch keine Bewertungen
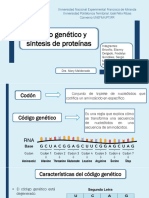
- Código Genético y Síntesis de ProteínasDokument15 SeitenCódigo Genético y Síntesis de ProteínasMariana100% (1)
- Procesos de Iniciación de Cadena Rezagada en La Replicación de ADN de EucariotasDokument3 SeitenProcesos de Iniciación de Cadena Rezagada en La Replicación de ADN de EucariotasAdriana Vanesa Tueros GuzmánNoch keine Bewertungen
- Bipiav9g 10Dokument9 SeitenBipiav9g 10Milton Edward Robles ArquerosNoch keine Bewertungen
- Aplicación de Bases Moelculares PDFDokument2 SeitenAplicación de Bases Moelculares PDFPao SotoNoch keine Bewertungen
- Presentación AcidosNucleicosDokument17 SeitenPresentación AcidosNucleicosEdith DanielNoch keine Bewertungen
- Biología (Adn y Arn)Dokument3 SeitenBiología (Adn y Arn)Lizbeth Erquinio bravoNoch keine Bewertungen
- Tarea 2 BioDokument4 SeitenTarea 2 Bioarodriguezb1005Noch keine Bewertungen
- Taller ADN Replicaciòn, Transcripción y TraducciònDokument8 SeitenTaller ADN Replicaciòn, Transcripción y TraducciònDarwin VelásquezNoch keine Bewertungen
- Protein Posttranslational Modifications - En.esDokument31 SeitenProtein Posttranslational Modifications - En.esJUAN PABLO CORREA ARROYAVENoch keine Bewertungen
- Tecnicas ADN-Aislam - Hibridacion.Dokument55 SeitenTecnicas ADN-Aislam - Hibridacion.Jean Rainel Yanez CabezasNoch keine Bewertungen
- 03 CiclosvirusDokument1 Seite03 Ciclosvirusapi-3700689Noch keine Bewertungen
- Final Bioquimica IiDokument26 SeitenFinal Bioquimica IiSofia GarciaNoch keine Bewertungen
- Adn y ArnDokument1 SeiteAdn y ArnManuel LaraNoch keine Bewertungen
- 2021 Motivacion LaboralDokument20 Seiten2021 Motivacion LaboralwaldirNoch keine Bewertungen
- Guia Didáctica Modelo CanvasDokument112 SeitenGuia Didáctica Modelo CanvasOscar Roberto Recinos Cañas100% (9)
- 530 Preguntas PSU Oficial RectificadoDokument85 Seiten530 Preguntas PSU Oficial RectificadoJessica Riquelme100% (2)
- Yo Soy Abundancia Patricia AnayaDokument220 SeitenYo Soy Abundancia Patricia AnayaJOctavioMuñozReyesNoch keine Bewertungen
- Yo Soy Abundancia Patricia AnayaDokument220 SeitenYo Soy Abundancia Patricia AnayaJOctavioMuñozReyesNoch keine Bewertungen
- RVM #235-2021-MineduDokument220 SeitenRVM #235-2021-MineduvilmaNoch keine Bewertungen
- Trabajos en Altura 07-08Dokument32 SeitenTrabajos en Altura 07-08waldirNoch keine Bewertungen
- Pauta - Planes - de - Negocio - Cafe - PROCOMPITE PDFDokument140 SeitenPauta - Planes - de - Negocio - Cafe - PROCOMPITE PDFDenisse Trudy Ingaroca FabianNoch keine Bewertungen
- Produ To Send o XenosDokument100 SeitenProdu To Send o XenosJavi Quispillo HuamánNoch keine Bewertungen
- Guia Tecnica de Viveros y Semilleros FrutalesDokument40 SeitenGuia Tecnica de Viveros y Semilleros FrutalesFabian Cruz R100% (1)
- Nectar de LimonDokument35 SeitenNectar de LimonwaldirNoch keine Bewertungen
- Proyecto de Mantenimiento de Vivero MunicipalDokument35 SeitenProyecto de Mantenimiento de Vivero MunicipalAyelinNoch keine Bewertungen
- Codex para Zumos (Jugos) y Néctares de FrutasDokument21 SeitenCodex para Zumos (Jugos) y Néctares de FrutasLuis Torres100% (1)
- Como Comenzar Un Negocio de Exportación PROMPERUDokument35 SeitenComo Comenzar Un Negocio de Exportación PROMPERUGabriela K. Vilca CruzNoch keine Bewertungen
- DESHIDRATACIONDokument140 SeitenDESHIDRATACIONLanders Salas RolandoNoch keine Bewertungen
- Encuesta de Residuos SolidosDokument3 SeitenEncuesta de Residuos SolidoswaldirNoch keine Bewertungen
- Modulo 1 - Tema 2. Tipos de Modelización Con LS-DYNA IDokument12 SeitenModulo 1 - Tema 2. Tipos de Modelización Con LS-DYNA IwaldirNoch keine Bewertungen
- Modulo 1 - Tema 2. Tipos de Modelización Con LS-DYNA I PDFDokument6 SeitenModulo 1 - Tema 2. Tipos de Modelización Con LS-DYNA I PDFwaldirNoch keine Bewertungen
- Colombia Aranceles y Politica ComercialDokument3 SeitenColombia Aranceles y Politica ComercialwaldirNoch keine Bewertungen
- Modulo 1 - Tema 1 - Introducción A LS - DYNADokument8 SeitenModulo 1 - Tema 1 - Introducción A LS - DYNAAnonymous siROwVWXNoch keine Bewertungen
- Modulo 1 - Tema 2. Tipos de Modelización Con LS-DYNA I PDFDokument6 SeitenModulo 1 - Tema 2. Tipos de Modelización Con LS-DYNA I PDFwaldirNoch keine Bewertungen
- Modelos de OligopolioDokument10 SeitenModelos de OligopolioCristian MoratayaNoch keine Bewertungen
- Separata Pulpas Nèctares, Merm Desh, Osmodes y Fruta ConfitadaDokument61 SeitenSeparata Pulpas Nèctares, Merm Desh, Osmodes y Fruta ConfitadaLuis MidWan100% (1)
- Técnicas de Procesado de CarnesDokument17 SeitenTécnicas de Procesado de CarnesDarwinVargasMantillaNoch keine Bewertungen
- Operaciones BancariasDokument26 SeitenOperaciones BancariaswaldirNoch keine Bewertungen
- Marco Conceptual de ImportacionesDokument54 SeitenMarco Conceptual de ImportacioneswaldirNoch keine Bewertungen
- Paper de Cianuracion AgitacionDokument8 SeitenPaper de Cianuracion AgitacionNano NandoNoch keine Bewertungen
- Argentina Aranceles y Politica ComercialDokument6 SeitenArgentina Aranceles y Politica ComercialwaldirNoch keine Bewertungen
- FAQ Mód1 EstadisticaDokument9 SeitenFAQ Mód1 EstadisticaJorshinio Salazar CastilloNoch keine Bewertungen
- NutricionDokument14 SeitenNutricionwayaveraNoch keine Bewertungen
- El PreformacionismoDokument4 SeitenEl PreformacionismoWonder DiazNoch keine Bewertungen
- Pci Ciencias NaturalesDokument34 SeitenPci Ciencias NaturalesEsperanza PomaNoch keine Bewertungen
- Taller de Sistema Circulatorio HumanoDokument6 SeitenTaller de Sistema Circulatorio HumanoCarol Dayan Chavarro SilvaNoch keine Bewertungen
- Sensacion y PercepcionDokument3 SeitenSensacion y PercepcionPiero BejaranoNoch keine Bewertungen
- Modelo de TripticoDokument2 SeitenModelo de TripticoVictor A. Figueroa Marcos0% (1)
- Manejo de Cepas Micro Biologic AsDokument12 SeitenManejo de Cepas Micro Biologic AsPatricia TorresNoch keine Bewertungen
- Ecogeneral MetapoblacionesDokument16 SeitenEcogeneral MetapoblacionesEnmanuel Valero0% (1)
- Brochurepdf AlteoDokument6 SeitenBrochurepdf AlteoD- AGROCOLNoch keine Bewertungen
- BiqouimicDokument6 SeitenBiqouimicBreynerChunquiSuarezNoch keine Bewertungen
- Ejercicios Turbidimetría y NefelometríaDokument2 SeitenEjercicios Turbidimetría y Nefelometríachorvo100% (1)
- 03) TaxonomíaDokument44 Seiten03) TaxonomíaAntonio ScarNoch keine Bewertungen
- CHerres Bermeo Jonathan David - Líquidos y ElectrolitosDokument3 SeitenCHerres Bermeo Jonathan David - Líquidos y ElectrolitosJonathan CherresNoch keine Bewertungen
- Antognazza Jorge - Aprendizaje Sin EstresDokument140 SeitenAntognazza Jorge - Aprendizaje Sin EstresCisco CanizNoch keine Bewertungen
- Neurona - WikipediaDokument7 SeitenNeurona - WikipediaVíctor MamaniNoch keine Bewertungen
- Planificación 1er Año Segundo LapsoDokument11 SeitenPlanificación 1er Año Segundo LapsocarNoch keine Bewertungen
- Utilidad ClínicaDokument9 SeitenUtilidad ClínicaMiguel ElreyNoch keine Bewertungen
- Sexaje de Aves Por PCRDokument6 SeitenSexaje de Aves Por PCRjoselyn.yumblaNoch keine Bewertungen
- Enfermedades Geneticas o Alteraciones Del Careotipo HumanoDokument16 SeitenEnfermedades Geneticas o Alteraciones Del Careotipo HumanoCrobanosky WujocaNoch keine Bewertungen
- Esporas y Su ImportaciaDokument43 SeitenEsporas y Su ImportaciaJavier RodriguezNoch keine Bewertungen
- Tema 22Dokument12 SeitenTema 22Oscar GomezNoch keine Bewertungen
- 4°M Biología ADN y ARN Prueba SumativaDokument5 Seiten4°M Biología ADN y ARN Prueba SumativaNataly Neira Guzmán100% (1)
- 7 Gastrotricos PDFDokument14 Seiten7 Gastrotricos PDFWILMAR SEBASTIAN FLECHAS SUAREZ100% (1)
- U-2 ProtozoariosDokument43 SeitenU-2 Protozoariosbubulubu37Noch keine Bewertungen
- E. Coli BLEEDokument9 SeitenE. Coli BLEEFernanda BastiasNoch keine Bewertungen
- Informe Microbiología N-3Dokument7 SeitenInforme Microbiología N-3Yanet HCNoch keine Bewertungen
- Atrofia MuscularDokument3 SeitenAtrofia MuscularCleidy Saavedra100% (1)
- OmicetesDokument5 SeitenOmicetesvengasusNoch keine Bewertungen
- Mapa Mental Educacion ProhibidaDokument7 SeitenMapa Mental Educacion ProhibidaDORIS VALLESNoch keine Bewertungen
- Manual Guardabosque VoluntarioDokument72 SeitenManual Guardabosque VoluntarioOscar Uchofen MenaNoch keine Bewertungen
- Adenosín DifosfatoDokument2 SeitenAdenosín DifosfatoBeto de la CruzNoch keine Bewertungen