Das könnte Ihnen auch gefallen
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryVon EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryBewertung: 3.5 von 5 Sternen3.5/5 (231)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Von EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Bewertung: 4.5 von 5 Sternen4.5/5 (121)
- Grit: The Power of Passion and PerseveranceVon EverandGrit: The Power of Passion and PerseveranceBewertung: 4 von 5 Sternen4/5 (588)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaVon EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaBewertung: 4.5 von 5 Sternen4.5/5 (266)
- The Little Book of Hygge: Danish Secrets to Happy LivingVon EverandThe Little Book of Hygge: Danish Secrets to Happy LivingBewertung: 3.5 von 5 Sternen3.5/5 (399)
- Never Split the Difference: Negotiating As If Your Life Depended On ItVon EverandNever Split the Difference: Negotiating As If Your Life Depended On ItBewertung: 4.5 von 5 Sternen4.5/5 (838)
- Shoe Dog: A Memoir by the Creator of NikeVon EverandShoe Dog: A Memoir by the Creator of NikeBewertung: 4.5 von 5 Sternen4.5/5 (537)
- The Emperor of All Maladies: A Biography of CancerVon EverandThe Emperor of All Maladies: A Biography of CancerBewertung: 4.5 von 5 Sternen4.5/5 (271)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeVon EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeBewertung: 4 von 5 Sternen4/5 (5794)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyVon EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyBewertung: 3.5 von 5 Sternen3.5/5 (2259)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersVon EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersBewertung: 4.5 von 5 Sternen4.5/5 (344)
- Team of Rivals: The Political Genius of Abraham LincolnVon EverandTeam of Rivals: The Political Genius of Abraham LincolnBewertung: 4.5 von 5 Sternen4.5/5 (234)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreVon EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreBewertung: 4 von 5 Sternen4/5 (1090)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceVon EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceBewertung: 4 von 5 Sternen4/5 (895)
- Her Body and Other Parties: StoriesVon EverandHer Body and Other Parties: StoriesBewertung: 4 von 5 Sternen4/5 (821)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureVon EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureBewertung: 4.5 von 5 Sternen4.5/5 (474)
- The Unwinding: An Inner History of the New AmericaVon EverandThe Unwinding: An Inner History of the New AmericaBewertung: 4 von 5 Sternen4/5 (45)
- BS7430 Earthing CalculationDokument14 SeitenBS7430 Earthing CalculationgyanNoch keine Bewertungen
- The Yellow House: A Memoir (2019 National Book Award Winner)Von EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Bewertung: 4 von 5 Sternen4/5 (98)
- On Fire: The (Burning) Case for a Green New DealVon EverandOn Fire: The (Burning) Case for a Green New DealBewertung: 4 von 5 Sternen4/5 (73)
- Catalog NeosetDokument173 SeitenCatalog NeosetCarmen Draghia100% (1)
- Daily Lesson Log (English)Dokument8 SeitenDaily Lesson Log (English)Julius Baldivino88% (8)
- The Role of Needs Analysis in Adult ESL Programme Design: Geoffrey BrindleyDokument16 SeitenThe Role of Needs Analysis in Adult ESL Programme Design: Geoffrey Brindleydeise krieser100% (2)
- Case Study III - MichelinDokument15 SeitenCase Study III - MichelinfreitzNoch keine Bewertungen
- Agenda - Meeting SLC (LT) - 27.06.2014 PDFDokument27 SeitenAgenda - Meeting SLC (LT) - 27.06.2014 PDFharshal1223Noch keine Bewertungen
- Group 2 - BSCE1 3 - Formal Lab Report#6 - CET 0122.1 11 2Dokument5 SeitenGroup 2 - BSCE1 3 - Formal Lab Report#6 - CET 0122.1 11 2John Eazer FranciscoNoch keine Bewertungen
- Torrent - WSCC - Windows System Control Center 7.0.5.7 Commercial (x64 x86) - TeamOS - Team OS - Your Only Destination To Custom OS !!Dokument5 SeitenTorrent - WSCC - Windows System Control Center 7.0.5.7 Commercial (x64 x86) - TeamOS - Team OS - Your Only Destination To Custom OS !!moustafaNoch keine Bewertungen
- RV900S - IB - Series 3Dokument28 SeitenRV900S - IB - Series 3GA LewisNoch keine Bewertungen
- Watershed Conservation of Benguet VisDokument2 SeitenWatershed Conservation of Benguet VisInnah Agito-RamosNoch keine Bewertungen
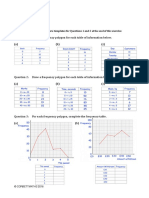
- Frequency Polygons pdf3Dokument7 SeitenFrequency Polygons pdf3Nevine El shendidyNoch keine Bewertungen
- Motive 27Tmx: Data SheetDokument2 SeitenMotive 27Tmx: Data SheetUlisesGómezNoch keine Bewertungen
- 240-56062705 RTV Silicone Rubber Insulator Coating and Shed Extender Supplier StandardDokument10 Seiten240-56062705 RTV Silicone Rubber Insulator Coating and Shed Extender Supplier StandardJane ChatsiriphatthanaNoch keine Bewertungen
- Company Profile PT. Geo Sriwijaya NusantaraDokument10 SeitenCompany Profile PT. Geo Sriwijaya NusantaraHazred Umar FathanNoch keine Bewertungen
- Sai Deepa Rock Drills: Unless Otherwise Specified ToleranceDokument1 SeiteSai Deepa Rock Drills: Unless Otherwise Specified ToleranceRavi BabaladiNoch keine Bewertungen
- GARCH (1,1) Models: Ruprecht-Karls-Universit at HeidelbergDokument42 SeitenGARCH (1,1) Models: Ruprecht-Karls-Universit at HeidelbergRanjan KumarNoch keine Bewertungen
- DICKSON KT800/802/803/804/856: Getting StartedDokument6 SeitenDICKSON KT800/802/803/804/856: Getting StartedkmpoulosNoch keine Bewertungen
- Answer:: Exercise-IDokument15 SeitenAnswer:: Exercise-IAishika NagNoch keine Bewertungen
- Bearing TypesDokument5 SeitenBearing TypesWayuNoch keine Bewertungen
- Handbook+for+Participants+ +GCC+TeenDokument59 SeitenHandbook+for+Participants+ +GCC+Teenchloe.2021164Noch keine Bewertungen
- Class I Water Well: DescriptionDokument10 SeitenClass I Water Well: DescriptionJavier Andrés Acevedo GarcíaNoch keine Bewertungen
- Time Table & Instruction For Candidate - Faculty of Sci & TechDokument3 SeitenTime Table & Instruction For Candidate - Faculty of Sci & TechDeepshikha Mehta joshiNoch keine Bewertungen
- AT ChapIDokument48 SeitenAT ChapIvigneshwaranbeNoch keine Bewertungen
- PLASSON UK July 2022 Price Catalogue v1Dokument74 SeitenPLASSON UK July 2022 Price Catalogue v1Jonathan Ninapaytan SanchezNoch keine Bewertungen
- Curriculum Improvement v2Dokument47 SeitenCurriculum Improvement v2Nica Lagrimas100% (1)
- FhryhfhfhDokument3 SeitenFhryhfhfhAffan AhmadNoch keine Bewertungen
- Gilbert Cell Design PDFDokument22 SeitenGilbert Cell Design PDFvysNoch keine Bewertungen
- STFC-2023 International E - conference-BITDokument6 SeitenSTFC-2023 International E - conference-BITRanilprabhu MNoch keine Bewertungen
- Changing Historical Perspectives On The Nazi DictatorshipDokument9 SeitenChanging Historical Perspectives On The Nazi Dictatorshipuploadimage666Noch keine Bewertungen
- Corometrics 170 Series BrochureDokument3 SeitenCorometrics 170 Series BrochureCesar MolanoNoch keine Bewertungen