Das könnte Ihnen auch gefallen
- Tipos de GalactosemiaDokument2 SeitenTipos de GalactosemiamelyNoch keine Bewertungen
- Metabolismo de GalactosaDokument12 SeitenMetabolismo de Galactosathankless_girlNoch keine Bewertungen
- GalactosemiaDokument18 SeitenGalactosemiaFelipeTepaleNoch keine Bewertungen
- Alteración Del Metabolismo de La GalactosaDokument12 SeitenAlteración Del Metabolismo de La GalactosaMariaIpaqNoch keine Bewertungen
- La Vía de Pentosa Fosfato 1Dokument9 SeitenLa Vía de Pentosa Fosfato 1Nirioky SanchezNoch keine Bewertungen
- GALACTOSEMIADokument13 SeitenGALACTOSEMIASalvador Cabrera CernNoch keine Bewertungen
- Bioquimica Equipo 4Dokument36 SeitenBioquimica Equipo 4Edna Quinto SantiagoNoch keine Bewertungen
- Galactosemia: el metabolismo de la galactosa y sus deficienciasDokument16 SeitenGalactosemia: el metabolismo de la galactosa y sus deficienciasJose Antonio GarciaNoch keine Bewertungen
- Bio Exposición MeDokument18 SeitenBio Exposición MeNATHALI DORIS PE�A TARQUINoch keine Bewertungen
- La galactosemia, una enfermedad hereditaria causada por deficiencia enzimáticaDokument23 SeitenLa galactosemia, una enfermedad hereditaria causada por deficiencia enzimáticaBrenda López HernándezNoch keine Bewertungen
- Cribado Neonatal.Dokument13 SeitenCribado Neonatal.Astrid IbarraNoch keine Bewertungen
- GluconeogénesisDokument7 SeitenGluconeogénesisroquehernandezmonseNoch keine Bewertungen
- GalactosemiaDokument8 SeitenGalactosemiaLucas RivasNoch keine Bewertungen
- S12 - INBORN ERRORS OF METABOLISM WITH HEPATOPATHY - En.esDokument16 SeitenS12 - INBORN ERRORS OF METABOLISM WITH HEPATOPATHY - En.esedwardNoch keine Bewertungen
- Alteraciones Del Metabolismo de La GalactosaDokument13 SeitenAlteraciones Del Metabolismo de La GalactosaRocio AlmanzaNoch keine Bewertungen
- Trabajos de Metabolismo de Los CarbohidratosDokument3 SeitenTrabajos de Metabolismo de Los CarbohidratosKarol MontañoNoch keine Bewertungen
- Metabolismo de Carbohidratos: Alteraciones en El Metabolismo de Fructosa y GalactosaDokument25 SeitenMetabolismo de Carbohidratos: Alteraciones en El Metabolismo de Fructosa y GalactosaxXxAbRaHaMxXxNoch keine Bewertungen
- GalactosemiaDokument15 SeitenGalactosemiaFrancisco Abad100% (1)
- Exposicion Grupo 5Dokument20 SeitenExposicion Grupo 5Kenia FragosoNoch keine Bewertungen
- GalactosemiaDokument10 SeitenGalactosemiaCsc Ana PaolaNoch keine Bewertungen
- Alteraciones Del Metabolismo de La FructosaDokument9 SeitenAlteraciones Del Metabolismo de La FructosaJaimes Rocha OscarNoch keine Bewertungen
- GlucolisisDokument61 SeitenGlucolisisJUAN SEBASTIAN CIFUENTES RUSSINoch keine Bewertungen
- Intolerancia hereditaria a la fructosaDokument5 SeitenIntolerancia hereditaria a la fructosaAlexandra Santa Cruz BellidoNoch keine Bewertungen
- GalactosemiaDokument2 SeitenGalactosemiaLuisa BonillaNoch keine Bewertungen
- El Metabolismo de La Fructosa y La Galactosa. Vías de Ingreso de Estos Sustratos en La Glicólisis. Errores Innatos Del Metabolismo A La Fructosa, Galactosa. Otros AzucaresDokument51 SeitenEl Metabolismo de La Fructosa y La Galactosa. Vías de Ingreso de Estos Sustratos en La Glicólisis. Errores Innatos Del Metabolismo A La Fructosa, Galactosa. Otros AzucaresaalarianNoch keine Bewertungen
- Seminario 2 BioquimicaDokument4 SeitenSeminario 2 BioquimicaQoqe Catalán DuránNoch keine Bewertungen
- Ejercicio 3Dokument2 SeitenEjercicio 3ELIZABETHNoch keine Bewertungen
- El metabolismo de la fructosa y sus enzimas claveDokument8 SeitenEl metabolismo de la fructosa y sus enzimas claveRafaella EscobedoNoch keine Bewertungen
- Galactosemia BioquiDokument3 SeitenGalactosemia BioquiDaviana MilanoNoch keine Bewertungen
- Galactosemia: desorden genético que impide digerir galactosaDokument9 SeitenGalactosemia: desorden genético que impide digerir galactosaLuis Gilberto MoncadaNoch keine Bewertungen
- GalactosemiaDokument3 SeitenGalactosemiaPaulinaNoch keine Bewertungen
- Vía de Las PentosasDokument22 SeitenVía de Las Pentosaspedroa8aNoch keine Bewertungen
- Defectos de La Gluconeogenesis ActualDokument6 SeitenDefectos de La Gluconeogenesis ActualAlexis RosarioNoch keine Bewertungen
- Intolerancia A La FructosaDokument7 SeitenIntolerancia A La FructosaJesús AvilezNoch keine Bewertungen
- Clases Nº08 Asistencia Al Usuario Con Patologias.Dokument7 SeitenClases Nº08 Asistencia Al Usuario Con Patologias.elvaNoch keine Bewertungen
- GalactosemiaDokument8 SeitenGalactosemiaANGEL ELIAS ALVAREZ GONZALEZNoch keine Bewertungen
- Trastornos MetabólicosDokument5 SeitenTrastornos Metabólicosapi-3697492100% (3)
- Galactosemia: enfermedad hereditariaDokument4 SeitenGalactosemia: enfermedad hereditariaGabriel Alfonso León Arango100% (1)
- Metabolismo de La Galactosa y La FructosaDokument18 SeitenMetabolismo de La Galactosa y La Fructosainternet Clicknet100% (1)
- Alteraciones en El Metabolismo de CarbohidratosDokument25 SeitenAlteraciones en El Metabolismo de CarbohidratosFernando MunaycoNoch keine Bewertungen
- ABPDokument9 SeitenABPmarcelaNoch keine Bewertungen
- FructosuriaDokument11 SeitenFructosuriaprincessargeNoch keine Bewertungen
- T-18 Metabolismo de Otras HexoxasDokument23 SeitenT-18 Metabolismo de Otras HexoxasDenis PeraltaNoch keine Bewertungen
- Ga LactosaDokument9 SeitenGa LactosaBella Stefanny Morales GuevaraNoch keine Bewertungen
- Metabolismo de La FructosaDokument9 SeitenMetabolismo de La Fructosapseudomedico011Noch keine Bewertungen
- GalactosemiaDokument3 SeitenGalactosemiaOsiris GarciaNoch keine Bewertungen
- Galactosemia: Definición, Causas, Signos, Síntomas, DX y TX, e Info. AdicionalDokument3 SeitenGalactosemia: Definición, Causas, Signos, Síntomas, DX y TX, e Info. Adicionalisrael ruiz fierroNoch keine Bewertungen
- Deficiencia de Fructosa 1-6-DifosfatasaDokument2 SeitenDeficiencia de Fructosa 1-6-Difosfatasajose perezNoch keine Bewertungen
- Metabolismo CarbohidratosDokument16 SeitenMetabolismo CarbohidratosJulioNoch keine Bewertungen
- Metabolismo GalactosaDokument2 SeitenMetabolismo GalactosaDaphneNoch keine Bewertungen
- Diagnóstico de Las Alteraciones Metabólica en Los CarbohidratosDokument9 SeitenDiagnóstico de Las Alteraciones Metabólica en Los CarbohidratosJuan luis Guerra acostaNoch keine Bewertungen
- CC GalactosemiaDokument4 SeitenCC GalactosemiaChris MondragonNoch keine Bewertungen
- Galactosemias y Fructosurias - 3CM9 - Eq.2Dokument20 SeitenGalactosemias y Fructosurias - 3CM9 - Eq.2Adriana HernándezNoch keine Bewertungen
- Galactosemia: Enfermedad metabólica por falta de enzimas que metabolizan la galactosaDokument4 SeitenGalactosemia: Enfermedad metabólica por falta de enzimas que metabolizan la galactosaKathya berenice Trujillo HernándezNoch keine Bewertungen
- GalactosemiaDokument7 SeitenGalactosemiaCoco Chavez IreneNoch keine Bewertungen
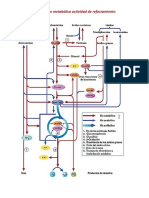
- Actividad de Reforzamiento Integracion MetabolicaDokument14 SeitenActividad de Reforzamiento Integracion MetabolicaJosselyn GaynorNoch keine Bewertungen
- Errores Congénitos Del MetabolismoDokument11 SeitenErrores Congénitos Del MetabolismoSergio Pratto Rillo100% (2)
- M3T4 - NeuropediatríaDokument9 SeitenM3T4 - NeuropediatríaAngela PortesNoch keine Bewertungen
- Alteraciones en El Metabolismo de CarbohidratosDokument18 SeitenAlteraciones en El Metabolismo de CarbohidratosMilagros Evelyn Aliaga EstupiñanNoch keine Bewertungen
- Soluciones para la Diabetes y la Hipoglucemia (Traducido): Cómo prevenirla y deshacerse de ella de forma natural, sin medicamentos pero adoptando un estilo de vida saludableVon EverandSoluciones para la Diabetes y la Hipoglucemia (Traducido): Cómo prevenirla y deshacerse de ella de forma natural, sin medicamentos pero adoptando un estilo de vida saludableNoch keine Bewertungen
- Quispe Molina Brayanm - OmicsDokument4 SeitenQuispe Molina Brayanm - OmicsBrayanm Quispe MolinaNoch keine Bewertungen
- Tratamiento de La Diabetes GestacionalDokument1 SeiteTratamiento de La Diabetes GestacionalBrayanm Quispe MolinaNoch keine Bewertungen
- Flujograma de Inocuidad HASSACDokument2 SeitenFlujograma de Inocuidad HASSACBrayanm Quispe MolinaNoch keine Bewertungen
- Reglamento Publicacion Revista Ciencia e InvestigaciónDokument2 SeitenReglamento Publicacion Revista Ciencia e InvestigaciónBrayanm Quispe MolinaNoch keine Bewertungen
- Modificadores Del PHDokument5 SeitenModificadores Del PHBrayanm Quispe MolinaNoch keine Bewertungen
- Metodologia AlcoholDokument3 SeitenMetodologia AlcoholBrayanm Quispe MolinaNoch keine Bewertungen
- Biosimilares-El Camino A ComenzadoDokument5 SeitenBiosimilares-El Camino A ComenzadoBrayanm Quispe MolinaNoch keine Bewertungen
- Infección UrinariaDokument8 SeitenInfección UrinariaBrayanm Quispe MolinaNoch keine Bewertungen
- Defectos en El TableteadoDokument7 SeitenDefectos en El TableteadoBrayanm Quispe MolinaNoch keine Bewertungen
- Analisis Cualitativo y Cuantitativo de Sertralina ClorhidratoDokument12 SeitenAnalisis Cualitativo y Cuantitativo de Sertralina ClorhidratoBrayanm Quispe Molina100% (3)
- Solicitud de PermisoDokument1 SeiteSolicitud de PermisoBrayanm Quispe MolinaNoch keine Bewertungen
- Factores Fisiopatologicos Que Alteran La Respuesta de UnDokument15 SeitenFactores Fisiopatologicos Que Alteran La Respuesta de UnBrayanm Quispe MolinaNoch keine Bewertungen
- Cuestionario Maleato de ClorferaminaDokument1 SeiteCuestionario Maleato de ClorferaminaBrayanm Quispe MolinaNoch keine Bewertungen
- Ramirez ReDokument112 SeitenRamirez ReBrayanm Quispe MolinaNoch keine Bewertungen
- Informe Identificacion Estructural de FarmacosDokument12 SeitenInforme Identificacion Estructural de FarmacosBrayanm Quispe MolinaNoch keine Bewertungen
- PovidoneDokument2 SeitenPovidoneBrayanm Quispe MolinaNoch keine Bewertungen
- Informe 3 de InstrumentalDokument3 SeitenInforme 3 de InstrumentalBrayanm Quispe MolinaNoch keine Bewertungen
- Análisis de Teoría de MediciónDokument4 SeitenAnálisis de Teoría de MediciónBrayanm Quispe MolinaNoch keine Bewertungen
- Cuestionario SertralinaDokument2 SeitenCuestionario SertralinaBrayanm Quispe MolinaNoch keine Bewertungen
- Proceso Formulacion Ciprofloxacino Handboock de FormulacionesDokument1 SeiteProceso Formulacion Ciprofloxacino Handboock de FormulacionesBrayanm Quispe MolinaNoch keine Bewertungen
- ME5 EcdiferencialesDokument31 SeitenME5 EcdiferencialesEdgar Copacondo MNoch keine Bewertungen
- Prevalencia de Interacciones Potenciales Evitables Entre Antidepresivos yDokument6 SeitenPrevalencia de Interacciones Potenciales Evitables Entre Antidepresivos yBrayanm Quispe MolinaNoch keine Bewertungen
- Efectos de La TemperaturaDokument3 SeitenEfectos de La TemperaturaBrayanm Quispe MolinaNoch keine Bewertungen
- En Qué Se Fundamenta La Extracción de Alcaloides enDokument1 SeiteEn Qué Se Fundamenta La Extracción de Alcaloides enBrayanm Quispe MolinaNoch keine Bewertungen
- Las Interacciones e Incompatibilidades de Excipientes Farmacéuticos Con Ingredientes Farmacéuticos Activos TraducidoDokument14 SeitenLas Interacciones e Incompatibilidades de Excipientes Farmacéuticos Con Ingredientes Farmacéuticos Activos TraducidoBrayanm Quispe Molina100% (4)
- Mezclado de LiquidosDokument11 SeitenMezclado de LiquidosBrayanm Quispe MolinaNoch keine Bewertungen
- Mecanismo de Reacción para La AcetanilidaDokument4 SeitenMecanismo de Reacción para La AcetanilidaBrayanm Quispe Molina0% (2)
- Agua para Inyectables o Agua Bidestilada 1Dokument10 SeitenAgua para Inyectables o Agua Bidestilada 1Brayanm Quispe MolinaNoch keine Bewertungen
- El Acondicionamiento Del AireDokument4 SeitenEl Acondicionamiento Del AireBrayanm Quispe MolinaNoch keine Bewertungen
- Materiales de PeluqueriaDokument2 SeitenMateriales de PeluquerialourdesNoch keine Bewertungen
- Métodos CromatográficosDokument10 SeitenMétodos CromatográficosALANNoch keine Bewertungen
- PRESUPUESTO ANALITICO - MOD N 02 FINAL - Camino de HerraduraDokument9 SeitenPRESUPUESTO ANALITICO - MOD N 02 FINAL - Camino de HerraduraWilber Paucar PaniuraNoch keine Bewertungen
- Benzoato de BenciloDokument16 SeitenBenzoato de BenciloEglee Cecilia Sanchez100% (1)
- Cronograma Bioquimica Ii F Medicina 2021-2Dokument23 SeitenCronograma Bioquimica Ii F Medicina 2021-2Danitza GutierrezNoch keine Bewertungen
- Acido FolicoDokument2 SeitenAcido FolicoJose María Caireta RuizNoch keine Bewertungen
- VIDEO 1: GLUCIDOS, LIPIDOS, PROTEINAS Y SU IMPORTANCIA EN LA NUTRICIÓN Y SALUDDokument7 SeitenVIDEO 1: GLUCIDOS, LIPIDOS, PROTEINAS Y SU IMPORTANCIA EN LA NUTRICIÓN Y SALUDdavid200520Noch keine Bewertungen
- Catálogo Teka 2017 2018 HornosDokument30 SeitenCatálogo Teka 2017 2018 HornosPau DuahruNoch keine Bewertungen
- Morinda Citrifolia (Noni)Dokument6 SeitenMorinda Citrifolia (Noni)Margarita Miranda RuizNoch keine Bewertungen
- Productos Ceramicos y Tabique en MuroDokument4 SeitenProductos Ceramicos y Tabique en MuroJuanNoch keine Bewertungen
- Cristalografía Formas CristalinasDokument15 SeitenCristalografía Formas CristalinasPiero Ramírez Torres100% (1)
- F04-P.LAB.01 V03 Cadena de Custodia de Toma de Muestra de AguaDokument1 SeiteF04-P.LAB.01 V03 Cadena de Custodia de Toma de Muestra de AguaLdynYanapaVelasquezNoch keine Bewertungen
- Soldadura-CompletoDokument65 SeitenSoldadura-CompletoGERLINoch keine Bewertungen
- Problemas RESUELTOS de PrecipitacionDokument17 SeitenProblemas RESUELTOS de PrecipitacionA Ver Pacheco100% (1)
- 8 Filtración Por MembranasDokument30 Seiten8 Filtración Por MembranasANGELA MAYERLY ACOSTA JARRONoch keine Bewertungen
- Elementos Complementarios de La Cadena de FríoDokument5 SeitenElementos Complementarios de La Cadena de Fríohaisenhair80% (5)
- Modelos de Fichas Tecnicas PDFDokument6 SeitenModelos de Fichas Tecnicas PDFFernando Coronado Mamani100% (1)
- Tema 10 Fisiologia XiomaDokument5 SeitenTema 10 Fisiologia Xiomafredy antonio de jesus nuñezNoch keine Bewertungen
- Equipos y Distribucion ElectricaDokument10 SeitenEquipos y Distribucion ElectricaEdison OrbeaNoch keine Bewertungen
- Ciclo de Krebs NuevoDokument33 SeitenCiclo de Krebs NuevoJoel PMNoch keine Bewertungen
- Mineria No Metalica - PaperDokument3 SeitenMineria No Metalica - PaperChuquimango Castrejon JhonatanNoch keine Bewertungen
- Historia de La Farmacia en ColombiaDokument3 SeitenHistoria de La Farmacia en Colombiabinsen lozano100% (2)
- 20) Tema6-Equilibrio de Formacion de Complejos (12 Sep)Dokument67 Seiten20) Tema6-Equilibrio de Formacion de Complejos (12 Sep)Matias Venegas MoraNoch keine Bewertungen
- Conversión de MataDokument28 SeitenConversión de MatafranckNoch keine Bewertungen
- Trabajo Práctico de Laboratorio N°Dokument5 SeitenTrabajo Práctico de Laboratorio N°sergioNoch keine Bewertungen
- Ficha Tec Hip Al 19%Dokument3 SeitenFicha Tec Hip Al 19%Calidad San Diego SESNoch keine Bewertungen
- Manual Plaguicidas P DocentesDokument64 SeitenManual Plaguicidas P DocentesOsvaldo De la IglesiaNoch keine Bewertungen
- Equi U1 Ea PargDokument9 SeitenEqui U1 Ea Pargpaolo antonio ruizNoch keine Bewertungen
- Libro Admon de MedicamentosDokument82 SeitenLibro Admon de Medicamentosjuan_mxNoch keine Bewertungen
- Control de Calidad de La LecheDokument15 SeitenControl de Calidad de La LecheLeiton Alvin Ramos VillenaNoch keine Bewertungen