Das könnte Ihnen auch gefallen
- Water Resources and Treatment, Wastewater and Water Quality ParametersDokument19 SeitenWater Resources and Treatment, Wastewater and Water Quality Parametersleslie_francisco_1Noch keine Bewertungen
- REED BED SYSTEM - ENS 301 NotesDokument11 SeitenREED BED SYSTEM - ENS 301 NotesDivya DiyaNoch keine Bewertungen
- CPP. Microproject PDF 2022Dokument11 SeitenCPP. Microproject PDF 2022Prathmesh ManusmareNoch keine Bewertungen
- Water Sources by Rey John C. BorjeDokument11 SeitenWater Sources by Rey John C. BorjeRey John BorjeNoch keine Bewertungen
- CHAPTER 2-Water PollutionDokument18 SeitenCHAPTER 2-Water PollutionkhairulhakamNoch keine Bewertungen
- Mary Grace BajaoDokument2 SeitenMary Grace BajaoShen CalotesNoch keine Bewertungen
- Design Aspects of Drinking Water PlantDokument32 SeitenDesign Aspects of Drinking Water PlantVenkatesh Kumar RamanujamNoch keine Bewertungen
- Environmental Engineering Assignment ON Desalination of WaterDokument17 SeitenEnvironmental Engineering Assignment ON Desalination of WaterAditya JalanNoch keine Bewertungen
- Presentation 1Dokument14 SeitenPresentation 1suhail4uNoch keine Bewertungen
- 1 s2.0 S2214714419316903 MainDokument18 Seiten1 s2.0 S2214714419316903 MainLuciano MagalhãesNoch keine Bewertungen
- 11 - Air and Water: Learning ObjectiveDokument4 Seiten11 - Air and Water: Learning ObjectiveAjay LakshmananNoch keine Bewertungen
- Waste Water TreatmentDokument8 SeitenWaste Water Treatmentdharshini ravichandranNoch keine Bewertungen
- Subject - : Environment Engineering - I (Water Supplying)Dokument11 SeitenSubject - : Environment Engineering - I (Water Supplying)Anuj KumarNoch keine Bewertungen
- HydrologyDokument7 SeitenHydrologyRohith GowdaNoch keine Bewertungen
- Gee 13Dokument10 SeitenGee 13Ana Marie AramayNoch keine Bewertungen
- WST Chapter 4 &5 HandoutDokument39 SeitenWST Chapter 4 &5 HandoutTadesse MegersaNoch keine Bewertungen
- Water Supply Use and ManagementDokument39 SeitenWater Supply Use and ManagementRoxette RoseteNoch keine Bewertungen
- Ecological Cycle of Water CycleDokument8 SeitenEcological Cycle of Water CycleKeth Jourly Misal PabillonNoch keine Bewertungen
- Desalination of Seawater by Reverse Osmosis Ro MethodDokument3 SeitenDesalination of Seawater by Reverse Osmosis Ro MethodInternational Journal of Innovative Science and Research TechnologyNoch keine Bewertungen
- Chapter 3 Dab 30102Dokument75 SeitenChapter 3 Dab 30102Muhammad Fazlan AliffNoch keine Bewertungen
- Water Filtration BackgroundDokument13 SeitenWater Filtration BackgroundDavidNoch keine Bewertungen
- Design Parameters of Sand Filtration Systems in Wastewater Treatment ProcessDokument9 SeitenDesign Parameters of Sand Filtration Systems in Wastewater Treatment ProcessMi doremiNoch keine Bewertungen
- Chapter-1: 1.1 Waste Water Treatment PlantDokument39 SeitenChapter-1: 1.1 Waste Water Treatment PlantKalyan Reddy AnuguNoch keine Bewertungen
- Oilfield Produced Water Management: Treatment, Reuse and DisposalDokument9 SeitenOilfield Produced Water Management: Treatment, Reuse and DisposalhafisjNoch keine Bewertungen
- Recycling of WaterDokument20 SeitenRecycling of Water10bcl002Noch keine Bewertungen
- AWA Water Journal Water in Mining Feb Edition 2014Dokument5 SeitenAWA Water Journal Water in Mining Feb Edition 2014Oktasari Dyah AnggrainiNoch keine Bewertungen
- Waste Water ParametersDokument5 SeitenWaste Water ParametersMay Joy OloyNoch keine Bewertungen
- Chapter ThreeDokument20 SeitenChapter ThreeKasozi Bateesa ShafickNoch keine Bewertungen
- Advance TreattDokument10 SeitenAdvance TreattwinkiNoch keine Bewertungen
- The Desalination UnitsDokument4 SeitenThe Desalination UnitssuinsasNoch keine Bewertungen
- Ppu QBDokument26 SeitenPpu QBAnonymous JDXbBDBNoch keine Bewertungen
- Potable Water Purification: Water Treatment Describes Those Industrial-Scale Processes Used To MakeDokument7 SeitenPotable Water Purification: Water Treatment Describes Those Industrial-Scale Processes Used To MakeKay RenNoch keine Bewertungen
- Conventional Water Treatment - Coagulation and Filtration - Safe Drinking Water FoundationDokument8 SeitenConventional Water Treatment - Coagulation and Filtration - Safe Drinking Water FoundationJimmy Hend KhratNoch keine Bewertungen
- Water Management1Dokument22 SeitenWater Management1Angelie Lape100% (1)
- Water Sewage Water Purification Using Solar Distillation SystemDokument37 SeitenWater Sewage Water Purification Using Solar Distillation Systempramo_dassNoch keine Bewertungen
- Institute of Graduate Studies: Romblon State University Odiongan, RomblonDokument4 SeitenInstitute of Graduate Studies: Romblon State University Odiongan, RomblonStevenzel Eala EstellaNoch keine Bewertungen
- Extraction of Freshwater and Energy From AtmosphereDokument14 SeitenExtraction of Freshwater and Energy From AtmosphereKate VentilacionNoch keine Bewertungen
- Seminar Report On Membrance TechnologyDokument32 SeitenSeminar Report On Membrance TechnologyAlok Shukla50% (2)
- PrintDokument10 SeitenPrintHasanNoch keine Bewertungen
- Water Purification Written ReportDokument10 SeitenWater Purification Written ReportMenric LunarNoch keine Bewertungen
- Water FiltrationDokument9 SeitenWater Filtrationmaliprathamesh13Noch keine Bewertungen
- Nanotechnology in Waste Water TreatmentDokument12 SeitenNanotechnology in Waste Water TreatmentLubna Amreen100% (1)
- CPI - Lesson 3ADokument24 SeitenCPI - Lesson 3AKarla Joy P. SucgangNoch keine Bewertungen
- Water Sources, Impurities, and Chemistry: The Planetary Water CycleDokument5 SeitenWater Sources, Impurities, and Chemistry: The Planetary Water CycleJoko SantosoNoch keine Bewertungen
- Water ReportDokument18 SeitenWater ReportMedwin Adam VillapandoNoch keine Bewertungen
- Dec 307 Enviromental EngineeringDokument7 SeitenDec 307 Enviromental Engineeringutachi93Noch keine Bewertungen
- CPI - Lesson 3Dokument16 SeitenCPI - Lesson 3Kim Tracey LadagaNoch keine Bewertungen
- Water PurificationDokument13 SeitenWater Purificationmister_no34Noch keine Bewertungen
- Unit II. Part - C. Water Resources ManagementDokument6 SeitenUnit II. Part - C. Water Resources ManagementAmruthRaj 'Ambu'Noch keine Bewertungen
- Water DesalinationDokument8 SeitenWater DesalinationMehwishNoch keine Bewertungen
- Treatment For Drinking Water ProductionDokument13 SeitenTreatment For Drinking Water ProductionmanjunathNoch keine Bewertungen
- Building Services IDokument43 SeitenBuilding Services IUtkarsh DwivediNoch keine Bewertungen
- New Water and Waste Water Treatment SystemDokument10 SeitenNew Water and Waste Water Treatment SystemSanthosh KumarNoch keine Bewertungen
- Chemistry 1Dokument79 SeitenChemistry 1malathi_vijayan417Noch keine Bewertungen
- 1 Nature and Importance of WaterDokument22 Seiten1 Nature and Importance of WaterGRAZIELLA CZARINA MARIE LABRADORNoch keine Bewertungen
- Reducing Mine Water RequirementsDokument12 SeitenReducing Mine Water RequirementsChalid MHNoch keine Bewertungen
- CHAPTER 3 - Water SupplyDokument76 SeitenCHAPTER 3 - Water SupplyMazliah Zainal Abidin100% (2)
- Microbiological Aspects of Pollution Control: Fundamental Aspects of Pollution Control and Environmental ScienceVon EverandMicrobiological Aspects of Pollution Control: Fundamental Aspects of Pollution Control and Environmental ScienceNoch keine Bewertungen
- BT 35 PDFDokument6 SeitenBT 35 PDFdalton2003Noch keine Bewertungen
- BT 25Dokument8 SeitenBT 25dalton2003Noch keine Bewertungen
- PBDokument6 SeitenPBdalton2003100% (1)
- Am Fsropro e Cell enDokument2 SeitenAm Fsropro e Cell endalton2003Noch keine Bewertungen
- PBDokument6 SeitenPBdalton2003100% (1)
- BT 58Dokument7 SeitenBT 58dalton2003Noch keine Bewertungen
- Owner/Operator Seminar Tuesday, February 7, 10:00a - 2:00p (Lunch Will Be Included)Dokument1 SeiteOwner/Operator Seminar Tuesday, February 7, 10:00a - 2:00p (Lunch Will Be Included)dalton2003Noch keine Bewertungen
- BT 31Dokument9 SeitenBT 31dalton2003Noch keine Bewertungen
- Flocon 285Dokument2 SeitenFlocon 285dalton2003Noch keine Bewertungen
- Water Treating Panel Discussion Monday, February 4, 2008 2:00p To 3:45pDokument4 SeitenWater Treating Panel Discussion Monday, February 4, 2008 2:00p To 3:45pdalton2003Noch keine Bewertungen
- Table To Play OutDokument1 SeiteTable To Play Outdalton2003Noch keine Bewertungen
- 2011 Seminar EducationDokument1 Seite2011 Seminar Educationdalton2003Noch keine Bewertungen
- 2006 Winter WT MinutesDokument2 Seiten2006 Winter WT Minutesdalton2003Noch keine Bewertungen
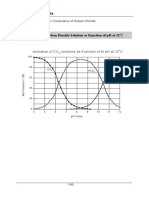
- Appendix: Ionization of CO Solutions As Function of TH PH at 25 CDokument1 SeiteAppendix: Ionization of CO Solutions As Function of TH PH at 25 Cdalton2003Noch keine Bewertungen
- CTSM 2 MSDS 1Dokument2 SeitenCTSM 2 MSDS 1dalton2003Noch keine Bewertungen
- Forugh Farrokhzad 2012 3Dokument71 SeitenForugh Farrokhzad 2012 3Eliana Galanda100% (1)
- Stabrom 909 MSDSDokument6 SeitenStabrom 909 MSDSdalton2003Noch keine Bewertungen
- Material Safety Data Sheet: U.S. Department of LaborDokument2 SeitenMaterial Safety Data Sheet: U.S. Department of Labordalton2003Noch keine Bewertungen
- Duboth OX Catalyzed - Product DataDokument1 SeiteDuboth OX Catalyzed - Product Datadalton2003Noch keine Bewertungen
- BWA Belcor 575 Replaces Molybdate USA WF 0Dokument2 SeitenBWA Belcor 575 Replaces Molybdate USA WF 0dalton2003Noch keine Bewertungen
- EDTMP Na5Dokument1 SeiteEDTMP Na5dalton2003Noch keine Bewertungen
- Aldehyde Keto. Ncert Book PDFDokument32 SeitenAldehyde Keto. Ncert Book PDFAshraf KhanNoch keine Bewertungen
- Naskah Drama Beauty and The BeastDokument39 SeitenNaskah Drama Beauty and The BeastAyu Rose75% (4)
- A Review On Battery Management System For Electric VehiclesDokument6 SeitenA Review On Battery Management System For Electric Vehiclesomkar chitnisNoch keine Bewertungen
- Clover by The RiverDokument24 SeitenClover by The RiverE. PoornimaNoch keine Bewertungen
- Craig - 4353 TX CobraDokument3 SeitenCraig - 4353 TX CobraJorge ContrerasNoch keine Bewertungen
- RediFlex Hoses Data SheetDokument2 SeitenRediFlex Hoses Data SheetNordson Adhesive Dispensing SystemsNoch keine Bewertungen
- Posttraumatic Stress Disorder (PTSD) and War-Related StressDokument56 SeitenPosttraumatic Stress Disorder (PTSD) and War-Related Stresshiggjp3Noch keine Bewertungen
- Principles of ForecastingDokument41 SeitenPrinciples of Forecastingrajeevseth100% (1)
- Homework 3rd SteelDokument4 SeitenHomework 3rd SteelPiseth HengNoch keine Bewertungen
- User'S Design Requirements For Single Chamber Pressure VesselsDokument8 SeitenUser'S Design Requirements For Single Chamber Pressure VesselspjsanchezmNoch keine Bewertungen
- MCQs Saudia Pharmacy Registration ExamDokument7 SeitenMCQs Saudia Pharmacy Registration ExamAli ButtNoch keine Bewertungen
- Basic Mechanical Engineering Btkit Dwarahat: Attempt All Questions. Q. 1. Attempt Any Four Parts: 5 X 4 20Dokument2 SeitenBasic Mechanical Engineering Btkit Dwarahat: Attempt All Questions. Q. 1. Attempt Any Four Parts: 5 X 4 20anadinath sharmaNoch keine Bewertungen
- Roger Ghanem, David Higdon, Houman Owhadi (Eds.) - Handbook of Uncertainty Quantification-Springer International Publishing (2017)Dokument2.035 SeitenRoger Ghanem, David Higdon, Houman Owhadi (Eds.) - Handbook of Uncertainty Quantification-Springer International Publishing (2017)Jaime Andres Cerda Garrido100% (1)
- Grade 11 Learning GuideDokument28 SeitenGrade 11 Learning GuideMary-Rose Casuyon100% (1)
- Hydraulics - MKM - DLX - Parts - Catalogue MAR 14 PDFDokument33 SeitenHydraulics - MKM - DLX - Parts - Catalogue MAR 14 PDFRS Rajib sarkerNoch keine Bewertungen
- PDF 1sz Fe Workshop Manual - CompressDokument2 SeitenPDF 1sz Fe Workshop Manual - CompressJose Luis Apaza Machaca75% (4)
- Class VII Half Yearly Maths, M.junaidDokument4 SeitenClass VII Half Yearly Maths, M.junaidmohd junaidNoch keine Bewertungen
- Agriculture Budget 2013-14Dokument33 SeitenAgriculture Budget 2013-14Ajay LimbasiyaNoch keine Bewertungen
- Msi MS 7529 Rev 1.1 PDFDokument33 SeitenMsi MS 7529 Rev 1.1 PDFMisael Alves67% (3)
- Related Literature BioplasticsDokument19 SeitenRelated Literature BioplasticsJames LimNoch keine Bewertungen
- CH 7. Pneumatic and HydroulicDokument20 SeitenCH 7. Pneumatic and HydroulicAbenezer Tasew100% (1)
- Scientific Method - AssessmentDokument13 SeitenScientific Method - AssessmentA.BensonNoch keine Bewertungen
- LEC - 19 - Task of Bitcoin MinersDokument36 SeitenLEC - 19 - Task of Bitcoin MinersKarunesh AnandNoch keine Bewertungen
- ENDOCRINE-BOARD REVIEW Dr. SchearDokument57 SeitenENDOCRINE-BOARD REVIEW Dr. SchearNayara PataroNoch keine Bewertungen
- Digital DividesDokument25 SeitenDigital DividesKumaraswamy ChannabasaiahNoch keine Bewertungen
- Comparative Study of Conventional and Generative Design ProcessDokument11 SeitenComparative Study of Conventional and Generative Design ProcessIJRASETPublicationsNoch keine Bewertungen
- Hydrostatics-Assignment 3: MPI td9Dokument2 SeitenHydrostatics-Assignment 3: MPI td9whoeverNoch keine Bewertungen
- Chapter 8 - Nervous ReviewerDokument18 SeitenChapter 8 - Nervous Reviewerchristian anchetaNoch keine Bewertungen
- International SubcontractingDokument2 SeitenInternational SubcontractingCatherine JohnsonNoch keine Bewertungen
- Exercises PDFDokument39 SeitenExercises PDF910220Noch keine Bewertungen