Das könnte Ihnen auch gefallen
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Von EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Bewertung: 4.5 von 5 Sternen4.5/5 (119)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaVon EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaBewertung: 4.5 von 5 Sternen4.5/5 (265)
- The Little Book of Hygge: Danish Secrets to Happy LivingVon EverandThe Little Book of Hygge: Danish Secrets to Happy LivingBewertung: 3.5 von 5 Sternen3.5/5 (399)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryVon EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryBewertung: 3.5 von 5 Sternen3.5/5 (231)
- Grit: The Power of Passion and PerseveranceVon EverandGrit: The Power of Passion and PerseveranceBewertung: 4 von 5 Sternen4/5 (587)
- Never Split the Difference: Negotiating As If Your Life Depended On ItVon EverandNever Split the Difference: Negotiating As If Your Life Depended On ItBewertung: 4.5 von 5 Sternen4.5/5 (838)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeVon EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeBewertung: 4 von 5 Sternen4/5 (5794)
- Team of Rivals: The Political Genius of Abraham LincolnVon EverandTeam of Rivals: The Political Genius of Abraham LincolnBewertung: 4.5 von 5 Sternen4.5/5 (234)
- Shoe Dog: A Memoir by the Creator of NikeVon EverandShoe Dog: A Memoir by the Creator of NikeBewertung: 4.5 von 5 Sternen4.5/5 (537)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyVon EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyBewertung: 3.5 von 5 Sternen3.5/5 (2219)
- The Emperor of All Maladies: A Biography of CancerVon EverandThe Emperor of All Maladies: A Biography of CancerBewertung: 4.5 von 5 Sternen4.5/5 (271)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreVon EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreBewertung: 4 von 5 Sternen4/5 (1090)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersVon EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersBewertung: 4.5 von 5 Sternen4.5/5 (344)
- Her Body and Other Parties: StoriesVon EverandHer Body and Other Parties: StoriesBewertung: 4 von 5 Sternen4/5 (821)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceVon EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceBewertung: 4 von 5 Sternen4/5 (894)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureVon EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureBewertung: 4.5 von 5 Sternen4.5/5 (474)
- The Unwinding: An Inner History of the New AmericaVon EverandThe Unwinding: An Inner History of the New AmericaBewertung: 4 von 5 Sternen4/5 (45)
- The Yellow House: A Memoir (2019 National Book Award Winner)Von EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Bewertung: 4 von 5 Sternen4/5 (98)
- On Fire: The (Burning) Case for a Green New DealVon EverandOn Fire: The (Burning) Case for a Green New DealBewertung: 4 von 5 Sternen4/5 (73)
- UWORLD Biostatistics Book Review PDFDokument93 SeitenUWORLD Biostatistics Book Review PDFসোমনাথ মহাপাত্র50% (2)
- Treatment of Recurrent Depression - John F. GredenDokument208 SeitenTreatment of Recurrent Depression - John F. GredenAlexAndraLuciaNoch keine Bewertungen
- Mercy Hospital Business PlanDokument14 SeitenMercy Hospital Business PlanNkem Joseph-PalmerNoch keine Bewertungen
- Medical AbbreviationsDokument4 SeitenMedical AbbreviationsNelly PaniaguaNoch keine Bewertungen
- British Pharmacopoeia 2022 BP 2022 - VOL 1Dokument1.440 SeitenBritish Pharmacopoeia 2022 BP 2022 - VOL 1Nguyễn Thu Trang100% (3)
- End of Life CareDokument80 SeitenEnd of Life CareRakesh MohanNoch keine Bewertungen
- Ectopic Pregnancy Nursing Care PlansDokument27 SeitenEctopic Pregnancy Nursing Care Plansviper7967880% (20)
- Brandtzaeg 1981Dokument14 SeitenBrandtzaeg 1981সোমনাথ মহাপাত্রNoch keine Bewertungen
- CBT 15 1029Dokument13 SeitenCBT 15 1029সোমনাথ মহাপাত্রNoch keine Bewertungen
- East Bank MapDokument1 SeiteEast Bank Mapসোমনাথ মহাপাত্রNoch keine Bewertungen
- My OwnDokument1 SeiteMy Ownসোমনাথ মহাপাত্রNoch keine Bewertungen
- Risk of 2nd Malig in NHLDokument11 SeitenRisk of 2nd Malig in NHLসোমনাথ মহাপাত্রNoch keine Bewertungen
- Globalizarea Nimicului - RitzerDokument27 SeitenGlobalizarea Nimicului - RitzerAlina PetreNoch keine Bewertungen
- Chapter 3 Haemoglobin Pattern Analysis: 3.1. Chromatographic Methods (For Hba Determination)Dokument27 SeitenChapter 3 Haemoglobin Pattern Analysis: 3.1. Chromatographic Methods (For Hba Determination)সোমনাথ মহাপাত্রNoch keine Bewertungen
- BD Biosciences Support - Protocols - Intracellular Staining of Human Red Blood CellsDokument1 SeiteBD Biosciences Support - Protocols - Intracellular Staining of Human Red Blood Cellsসোমনাথ মহাপাত্রNoch keine Bewertungen
- 1201020895PL IMillerDokument26 Seiten1201020895PL IMillerসোমনাথ মহাপাত্রNoch keine Bewertungen
- Account Statement As of 04-05-2020 19:48:27 GMT +0530Dokument11 SeitenAccount Statement As of 04-05-2020 19:48:27 GMT +0530সোমনাথ মহাপাত্রNoch keine Bewertungen
- Immune Recognition of Self Nucleic Acids Driven by Endogenous Antimicrobial Peptides: Role in AutoimmunityDokument176 SeitenImmune Recognition of Self Nucleic Acids Driven by Endogenous Antimicrobial Peptides: Role in Autoimmunityসোমনাথ মহাপাত্রNoch keine Bewertungen
- 149 FullDokument10 Seiten149 Fullসোমনাথ মহাপাত্রNoch keine Bewertungen
- DHR-ICMR Workshop Funding ApplicationDokument3 SeitenDHR-ICMR Workshop Funding Applicationসোমনাথ মহাপাত্রNoch keine Bewertungen
- Chap1 - Units, Dimensions & Vectors PDFDokument61 SeitenChap1 - Units, Dimensions & Vectors PDFসোমনাথ মহাপাত্রNoch keine Bewertungen
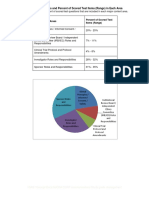
- Five Content Areas and Percent of Scored Test Items (Range) in Each AreaDokument5 SeitenFive Content Areas and Percent of Scored Test Items (Range) in Each Areaসোমনাথ মহাপাত্রNoch keine Bewertungen
- Cell-Cell Interactions: Concept OutlineDokument18 SeitenCell-Cell Interactions: Concept Outlineসোমনাথ মহাপাত্রNoch keine Bewertungen
- DHR-ICMR Workshop Funding ApplicationDokument3 SeitenDHR-ICMR Workshop Funding Applicationসোমনাথ মহাপাত্রNoch keine Bewertungen
- Result Sheet PDFDokument41 SeitenResult Sheet PDFManohar ManuNoch keine Bewertungen
- Icmr Talent FormDokument2 SeitenIcmr Talent Formসোমনাথ মহাপাত্রNoch keine Bewertungen
- EBv Neg Epidemiology Leblond JCO.1998.16.6Dokument8 SeitenEBv Neg Epidemiology Leblond JCO.1998.16.6সোমনাথ মহাপাত্রNoch keine Bewertungen
- Boards and Beyound Step 2 Cs PalpitationsDokument4 SeitenBoards and Beyound Step 2 Cs Palpitationsসোমনাথ মহাপাত্রNoch keine Bewertungen
- Griffin Observership ApplicationDokument7 SeitenGriffin Observership Applicationসোমনাথ মহাপাত্রNoch keine Bewertungen
- Chemo For HematologyDokument1 SeiteChemo For Hematologyp monica aneeshaNoch keine Bewertungen
- Anxiety Disorders Treatment ProtocolDokument25 SeitenAnxiety Disorders Treatment Protocolসোমনাথ মহাপাত্রNoch keine Bewertungen
- 1 This Is System WiseDokument1 Seite1 This Is System Wiseসোমনাথ মহাপাত্রNoch keine Bewertungen
- Chemo For HematologyDokument1 SeiteChemo For Hematologyp monica aneeshaNoch keine Bewertungen
- IMG Observership PolicyDokument3 SeitenIMG Observership PolicyNilay BhattNoch keine Bewertungen
- Cs Info ManualDokument20 SeitenCs Info Manualসোমনাথ মহাপাত্রNoch keine Bewertungen
- DispesingDokument36 SeitenDispesingLupus BoyssNoch keine Bewertungen
- Epi#3Dokument19 SeitenEpi#3JNoch keine Bewertungen
- Case Report: Diastema Closure in Anterior Teeth Using A Posterior MatrixDokument7 SeitenCase Report: Diastema Closure in Anterior Teeth Using A Posterior MatrixJing XueNoch keine Bewertungen
- Care of Vulnarable PatientsDokument30 SeitenCare of Vulnarable PatientsMariya DantisNoch keine Bewertungen
- Standards For Good Pharmacy Practice - A Comparative AnalysisDokument6 SeitenStandards For Good Pharmacy Practice - A Comparative AnalysisClaudia NovăceanNoch keine Bewertungen
- HRM Case Study-PatelDokument11 SeitenHRM Case Study-PatelHumaira ShafiqNoch keine Bewertungen
- FCD Ethical and Legal IssuesDokument6 SeitenFCD Ethical and Legal Issuesnicholas51292Noch keine Bewertungen
- Ef205 Unit 2 Assignment 1Dokument5 SeitenEf205 Unit 2 Assignment 1api-601333476Noch keine Bewertungen
- Pharmacy Intravena Admixture Services (Pivas) : IV - Admixture Handling CytotoxicDokument32 SeitenPharmacy Intravena Admixture Services (Pivas) : IV - Admixture Handling CytotoxicintanNoch keine Bewertungen
- DY - DM&HO Daily Empty ReportDokument1 SeiteDY - DM&HO Daily Empty Reportphc kallumarriNoch keine Bewertungen
- List of Empanelled HCOs-Jaipur As On 20 December 2022Dokument24 SeitenList of Empanelled HCOs-Jaipur As On 20 December 2022SATYAJIT PALNoch keine Bewertungen
- Ethics in Midwifery PowerpointDokument24 SeitenEthics in Midwifery PowerpointEden NatividadNoch keine Bewertungen
- Assessment of Insulin Injection Practice of Nurses Working inDokument7 SeitenAssessment of Insulin Injection Practice of Nurses Working inBheru LalNoch keine Bewertungen
- Flowable Wound Matrix Outpatient Reimbursement Summary 2019Dokument4 SeitenFlowable Wound Matrix Outpatient Reimbursement Summary 2019Bob RiouxNoch keine Bewertungen
- Pa Tas Database (Conso)Dokument40 SeitenPa Tas Database (Conso)AlbeldaArnaldoNoch keine Bewertungen
- FMEC ProgramDokument25 SeitenFMEC ProgramVasuNoch keine Bewertungen
- Lic 602 ADokument6 SeitenLic 602 ALm RappeportNoch keine Bewertungen
- CHHS Clinical Handover ProcedureDokument16 SeitenCHHS Clinical Handover ProcedureSiu Andrea100% (1)
- Evwoman M00o03Dokument33 SeitenEvwoman M00o03Nader AlsheikhNoch keine Bewertungen
- Ssa 3368Dokument14 SeitenSsa 3368JonOstiguyNoch keine Bewertungen
- Chapter 5Dokument42 SeitenChapter 5Yuuki Chitose (tai-kun)Noch keine Bewertungen
- Aluminium Foil For Smoking DrugsDokument6 SeitenAluminium Foil For Smoking Drugscamille64Noch keine Bewertungen
- General DataDokument6 SeitenGeneral DataGilbert Allen MateoNoch keine Bewertungen
- Social Work - Lesson 8Dokument51 SeitenSocial Work - Lesson 8ARLETH FOLLERO100% (1)
- Cardiotocography Trace Pattern Evaluation Using MATLAB ProgramDokument6 SeitenCardiotocography Trace Pattern Evaluation Using MATLAB ProgramdianaNoch keine Bewertungen