Das könnte Ihnen auch gefallen
- Preliminary Specifications: Programmed Data Processor Model Three (PDP-3) October, 1960Von EverandPreliminary Specifications: Programmed Data Processor Model Three (PDP-3) October, 1960Noch keine Bewertungen
- HISAT2Dokument35 SeitenHISAT2Jelena Nađ100% (1)
- Propeller Programming: Using Assembler, Spin, and CVon EverandPropeller Programming: Using Assembler, Spin, and CNoch keine Bewertungen
- 34 Fastp An UltraDokument7 Seiten34 Fastp An UltraResul KıymazNoch keine Bewertungen
- Genomics Dataset: Prof. Devdatt Dubhashi & Dr. Mukund DeshpandeDokument11 SeitenGenomics Dataset: Prof. Devdatt Dubhashi & Dr. Mukund DeshpandeFrank IssNoch keine Bewertungen
- In Silico Genome Analysis-Inderjit (SoAB)Dokument5 SeitenIn Silico Genome Analysis-Inderjit (SoAB)tango0385Noch keine Bewertungen
- I R Assignment 1Dokument2 SeitenI R Assignment 1Arghya AdhyaNoch keine Bewertungen
- xwdgdswuhpryhvdgdswhu Vhtxhqfhviurpkljkwkurxjksxw VhtxhqflqjuhdgvDokument3 Seitenxwdgdswuhpryhvdgdswhu Vhtxhqfhviurpkljkwkurxjksxw VhtxhqflqjuhdgvOSCAR ALEXIS QUINTERO L�PEZNoch keine Bewertungen
- NGS ToolsFormats r1 BDGDokument32 SeitenNGS ToolsFormats r1 BDGRangga K NegaraNoch keine Bewertungen
- Analyzing of Pseudo-Ring Memory Self-Testing Schemes With AlgorithmsDokument8 SeitenAnalyzing of Pseudo-Ring Memory Self-Testing Schemes With AlgorithmsijdpsNoch keine Bewertungen
- Chaos-Based Bitwise Dynamical Pseudorandom Number Generator On FPGADokument4 SeitenChaos-Based Bitwise Dynamical Pseudorandom Number Generator On FPGATammy SguizzattoNoch keine Bewertungen
- Assignment 1Dokument2 SeitenAssignment 1Gourab PatroNoch keine Bewertungen
- Reliability Literature Review Assignment 3Dokument4 SeitenReliability Literature Review Assignment 3TIVIYAH MOGANNoch keine Bewertungen
- Brouwer1998 Chapter MythsAndFactsAboutTheEfficient PDFDokument15 SeitenBrouwer1998 Chapter MythsAndFactsAboutTheEfficient PDFrubaNoch keine Bewertungen
- Operating Systems (04JEZOQ)Dokument2 SeitenOperating Systems (04JEZOQ)Mustafa LulajNoch keine Bewertungen
- PTRAJ and CPPTRAJ: Software For Processing and Analysis of Molecular Dynamics Trajectory DataDokument12 SeitenPTRAJ and CPPTRAJ: Software For Processing and Analysis of Molecular Dynamics Trajectory DatasueNoch keine Bewertungen
- Modelo Estadístico Sobre El Frpsruwdplhqwrgho Wkurxjksxw en Uhghv/$1Vreuhwhfqrorjtd Srzhuolqh CommunicationsDokument16 SeitenModelo Estadístico Sobre El Frpsruwdplhqwrgho Wkurxjksxw en Uhghv/$1Vreuhwhfqrorjtd Srzhuolqh CommunicationsJessica Asitimbay ZuritaNoch keine Bewertungen
- UNIT-5: Department of Computer Science & EngineeringDokument25 SeitenUNIT-5: Department of Computer Science & Engineeringrosh benNoch keine Bewertungen
- Hkbu Thesis FormatDokument5 SeitenHkbu Thesis Formatihbjtphig100% (2)
- Lect 1 IntroDokument61 SeitenLect 1 IntroMohamed AkelNoch keine Bewertungen
- SlideDokument14 SeitenSlideapi-3845765100% (2)
- Slide1Dokument15 SeitenSlide1api-3845765100% (2)
- Random 123 SC 11Dokument12 SeitenRandom 123 SC 11Abed MomaniNoch keine Bewertungen
- Implementation of Dynamic Level Scheduling Algorithm Using Genetic OperatorsDokument5 SeitenImplementation of Dynamic Level Scheduling Algorithm Using Genetic OperatorsInternational Journal of Application or Innovation in Engineering & ManagementNoch keine Bewertungen
- Icimp 2016 2 30 30033Dokument7 SeitenIcimp 2016 2 30 30033Bruno RekowskyNoch keine Bewertungen
- BY:-Shruti Sachdeva 100905090: Thapar UniversityDokument54 SeitenBY:-Shruti Sachdeva 100905090: Thapar UniversityShruti SachdevaNoch keine Bewertungen
- CD Module2 16 03 23 PDFDokument36 SeitenCD Module2 16 03 23 PDFSouvik DasNoch keine Bewertungen
- Primr DesignDokument57 SeitenPrimr DesignSunilNoch keine Bewertungen
- FPGA Based Parallel Computation Techniques For Bioinformatics ApplicationsDokument5 SeitenFPGA Based Parallel Computation Techniques For Bioinformatics ApplicationsHugo ViníciusNoch keine Bewertungen
- Timing Analysis in Physical DesignDokument32 SeitenTiming Analysis in Physical Designgoud.mahesh0584269100% (2)
- R: A Reconfigurable Atomic Memory Service For Dynamic NetworksDokument18 SeitenR: A Reconfigurable Atomic Memory Service For Dynamic NetworksRoop StrongNoch keine Bewertungen
- SynTest Tutorial TSDokument1 SeiteSynTest Tutorial TSkapilhaliNoch keine Bewertungen
- Journal of Computational Physics: Y. Lin, F. Wang, B. LiuDokument11 SeitenJournal of Computational Physics: Y. Lin, F. Wang, B. LiupovNoch keine Bewertungen
- TMP DA0 DDokument13 SeitenTMP DA0 DFrontiersNoch keine Bewertungen
- 2016 Arpn Jeas Peak SocDokument9 Seiten2016 Arpn Jeas Peak SocNHI NGUYỄN HƯƠNGNoch keine Bewertungen
- D002.Lab Manual Laboratory Practice VDokument39 SeitenD002.Lab Manual Laboratory Practice Vtanay.bhor3Noch keine Bewertungen
- Department of Information Technology Assignment No. 2 TITLE: Implement Multithreading For Matrix Multiplication Using Pthreads. ObjectiveDokument6 SeitenDepartment of Information Technology Assignment No. 2 TITLE: Implement Multithreading For Matrix Multiplication Using Pthreads. ObjectiveDipali PatilNoch keine Bewertungen
- UPCOTDokument7 SeitenUPCOTArun UohNoch keine Bewertungen
- Thesis Reportt 2Dokument85 SeitenThesis Reportt 2MianHammadNazir100% (1)
- Parallel Libraries and Parallel I/O: John UrbanicDokument45 SeitenParallel Libraries and Parallel I/O: John UrbanicarjunNoch keine Bewertungen
- First LectureDokument89 SeitenFirst LectureMohamed HasanNoch keine Bewertungen
- Titanic: Mohit Kothari Roger Tanuatmadja Gautam AkiwateDokument18 SeitenTitanic: Mohit Kothari Roger Tanuatmadja Gautam AkiwatezarthonNoch keine Bewertungen
- WARP3D 17.4.0 Manual Updated June 5 2013Dokument484 SeitenWARP3D 17.4.0 Manual Updated June 5 2013marc53042Noch keine Bewertungen
- Routing Protocol ComparisonDokument8 SeitenRouting Protocol ComparisonBabar SaeedNoch keine Bewertungen
- File Structures Lab: Laboratory ManualDokument65 SeitenFile Structures Lab: Laboratory ManualANAGHANoch keine Bewertungen
- Analysis of RIP, OSPF, and EIGRP Routing": Synopsis ON "Dokument9 SeitenAnalysis of RIP, OSPF, and EIGRP Routing": Synopsis ON "Đěěpâķ ŠîňğhNoch keine Bewertungen
- Sanskrit Tag-Sets and Part-Of-Speech Tagging Methods - A SurveyDokument6 SeitenSanskrit Tag-Sets and Part-Of-Speech Tagging Methods - A Surveysujal1310Noch keine Bewertungen
- J.D. Opdyke - Permutation Tests (And Sampling Without Replacement) Orders of Magnitude Faster Using SAS - ScribeDokument40 SeitenJ.D. Opdyke - Permutation Tests (And Sampling Without Replacement) Orders of Magnitude Faster Using SAS - ScribejdopdykeNoch keine Bewertungen
- UpdatedbibliographyDokument5 SeitenUpdatedbibliographyapi-298940823Noch keine Bewertungen
- A "How To" Tutorial On Logic Analyzer Basics For Digital DesignDokument4 SeitenA "How To" Tutorial On Logic Analyzer Basics For Digital DesignshinojoseNoch keine Bewertungen
- HHS Public Access: Ballgown Bridges The Gap Between Transcriptome Assembly and Expression AnalysisDokument9 SeitenHHS Public Access: Ballgown Bridges The Gap Between Transcriptome Assembly and Expression AnalysisranjanmanishblueNoch keine Bewertungen
- OpenmpDokument21 SeitenOpenmpMark VeltzerNoch keine Bewertungen
- Parallel FPGA-based All-Pairs Shortest-Paths in A Directed GraphDokument10 SeitenParallel FPGA-based All-Pairs Shortest-Paths in A Directed Graphprakash_oneNoch keine Bewertungen
- Artificial Intelligence - Assignment 3Dokument11 SeitenArtificial Intelligence - Assignment 3Pankhuri BhatnagarNoch keine Bewertungen
- DAA R19 - All UnitsDokument219 SeitenDAA R19 - All Unitspujitha akumallaNoch keine Bewertungen
- 11.1 EtherChannelDokument3 Seiten11.1 EtherChannelamit_post2000Noch keine Bewertungen
- Week 2-3 - Analysis of AlgorithmsDokument7 SeitenWeek 2-3 - Analysis of AlgorithmsXuân Dương VươngNoch keine Bewertungen
- Parallel Performance Study of Monte Carlo Photon Transport Code On Shared-, Distributed-, and Distributed-Shared-Memory ArchitecturesDokument7 SeitenParallel Performance Study of Monte Carlo Photon Transport Code On Shared-, Distributed-, and Distributed-Shared-Memory Architecturesvermashanu89Noch keine Bewertungen
- SynopsisDokument13 SeitenSynopsisRangrez shaibazNoch keine Bewertungen
- Implementation and Analysis of An Error DetectionDokument7 SeitenImplementation and Analysis of An Error DetectioninfinitywaysalwaysNoch keine Bewertungen
- Unit 1 Java FundamentalsDokument51 SeitenUnit 1 Java Fundamentalsanshulvyas23Noch keine Bewertungen
- Exception Handling in JavaDokument2 SeitenException Handling in Javaanshulvyas23Noch keine Bewertungen
- MOSAIKSOAPDokument11 SeitenMOSAIKSOAPanshulvyas23Noch keine Bewertungen
- Why Do We Study Theory of Computation ?Dokument36 SeitenWhy Do We Study Theory of Computation ?anshulvyas23Noch keine Bewertungen
- MpiDokument30 SeitenMpianshulvyas23Noch keine Bewertungen
- DPC Principal 2015 16 28042015Dokument332 SeitenDPC Principal 2015 16 28042015anshulvyas23Noch keine Bewertungen
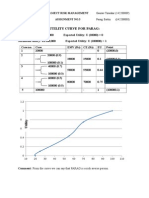
- Utility Curve For ParagDokument2 SeitenUtility Curve For Paraganshulvyas23Noch keine Bewertungen
- HomeworkDokument5 SeitenHomeworkvgciasenNoch keine Bewertungen
- HLP Action PlanDokument75 SeitenHLP Action PlanThiago GueirosNoch keine Bewertungen
- Biophysics SyllabusDokument2 SeitenBiophysics SyllabusKamlesh SahuNoch keine Bewertungen
- Handout - Cell Transport Review WorksheetDokument4 SeitenHandout - Cell Transport Review Worksheetapi-502781581Noch keine Bewertungen
- Tugasan Epraktikal XBDM4103 Mei 2021Dokument6 SeitenTugasan Epraktikal XBDM4103 Mei 2021norfilzahNoch keine Bewertungen
- Updated CV - JyotiDokument2 SeitenUpdated CV - Jyotigablu babluNoch keine Bewertungen
- Marker Assisted Selection (MAS)Dokument57 SeitenMarker Assisted Selection (MAS)teledaneNoch keine Bewertungen
- Anti-Microbial Activity of Cassia Tora Leaves and Stems Crude ExtractDokument4 SeitenAnti-Microbial Activity of Cassia Tora Leaves and Stems Crude ExtractHelixNoch keine Bewertungen
- 2015 Mobile Dna IIIDokument1.346 Seiten2015 Mobile Dna IIIMauro Ortiz100% (2)
- Us20120251502a1 PDFDokument117 SeitenUs20120251502a1 PDFFernando CabreraNoch keine Bewertungen
- Buku EvolusiDokument446 SeitenBuku Evolusigian septhayudi100% (1)
- Principles of Drug Testing TechnologyDokument2 SeitenPrinciples of Drug Testing Technologydrugtestsdirect100% (1)
- Cell CycleDokument117 SeitenCell CyclePahel AminNoch keine Bewertungen
- Molecular Biology of The Cell 5th Edition Alberts Test BankDokument13 SeitenMolecular Biology of The Cell 5th Edition Alberts Test Bankodettedieupmx23m100% (31)
- Lab 3 - Smear Preparation Simple Staining and Gram StainingDokument29 SeitenLab 3 - Smear Preparation Simple Staining and Gram StainingAyman ElkenawyNoch keine Bewertungen
- BotanyDokument48 SeitenBotanyVenky GVNoch keine Bewertungen
- Book Real-Time PCRDokument343 SeitenBook Real-Time PCRWida Salupi100% (1)
- Photosynthesis Inihibitors Review MyDokument2 SeitenPhotosynthesis Inihibitors Review Myapi-521781723Noch keine Bewertungen
- Malaysian Pharmaceutical Industry: Opportunities and ChallengesDokument8 SeitenMalaysian Pharmaceutical Industry: Opportunities and ChallengesChoko Ema SakuraNoch keine Bewertungen
- Bok:978 3 642 37922 2Dokument615 SeitenBok:978 3 642 37922 2atilio martinezNoch keine Bewertungen
- Department of Molecular Biology: Test Name Result Unit Bio. Ref. Range MethodDokument1 SeiteDepartment of Molecular Biology: Test Name Result Unit Bio. Ref. Range MethodRam TholetyNoch keine Bewertungen
- Haa Nipa Question Bank (Proposed) - FinalDokument3 SeitenHaa Nipa Question Bank (Proposed) - FinalZahidul HassanNoch keine Bewertungen
- Definition of Cell CultureDokument36 SeitenDefinition of Cell CultureGladys AilingNoch keine Bewertungen
- Developing A Sars-Cov 2 Antigen Test Using Engineered A Nity ProteinsDokument13 SeitenDeveloping A Sars-Cov 2 Antigen Test Using Engineered A Nity ProteinsKalyan KarumanchiNoch keine Bewertungen
- CVDokument4 SeitenCVapi-504844041Noch keine Bewertungen
- Data Monkey TutorialDokument31 SeitenData Monkey TutorialRebriarina HapsariNoch keine Bewertungen
- The Cell Cycle ScriptDokument5 SeitenThe Cell Cycle ScriptAlexNoch keine Bewertungen
- The Role of Clinical Pharmacist in Pharmacovigilance and Drug Safety in Teritiary Care Teaching HospitalDokument11 SeitenThe Role of Clinical Pharmacist in Pharmacovigilance and Drug Safety in Teritiary Care Teaching HospitalBaru Chandrasekhar RaoNoch keine Bewertungen
- Unit 2. Macromolecules of The Life and Their ImportanceDokument12 SeitenUnit 2. Macromolecules of The Life and Their ImportanceSherif AliNoch keine Bewertungen
- SUBHADIPA MAJUMDER2022-07-22Cell PotencyDokument2 SeitenSUBHADIPA MAJUMDER2022-07-22Cell PotencySuvNoch keine Bewertungen