Das könnte Ihnen auch gefallen
- Shoe Dog: A Memoir by the Creator of NikeVon EverandShoe Dog: A Memoir by the Creator of NikeBewertung: 4.5 von 5 Sternen4.5/5 (537)
- Grit: The Power of Passion and PerseveranceVon EverandGrit: The Power of Passion and PerseveranceBewertung: 4 von 5 Sternen4/5 (587)
- Kim Et Al. - 2011 - Hyperexpansion of GAA Repeats Affects Post-InitiatDokument12 SeitenKim Et Al. - 2011 - Hyperexpansion of GAA Repeats Affects Post-Initiatfidel muñozNoch keine Bewertungen
- Ashby How To Write A PaperDokument47 SeitenAshby How To Write A PaperGauri RanadiveNoch keine Bewertungen
- Chapdelaine Et Al. - 2013 - A Potential New Therapeutic Approach For FriedreicDokument9 SeitenChapdelaine Et Al. - 2013 - A Potential New Therapeutic Approach For Friedreicfidel muñozNoch keine Bewertungen
- Somatic MutationsDokument10 SeitenSomatic Mutationsfidel muñozNoch keine Bewertungen
- Chutake Et Al. - 2015 - FXN Promoter Silencing in The Humanized Mouse ModeDokument12 SeitenChutake Et Al. - 2015 - FXN Promoter Silencing in The Humanized Mouse Modefidel muñozNoch keine Bewertungen
- Koeppen Et Al. - 2012 - Friedreich's Ataxia Causes Redistribution of IronDokument16 SeitenKoeppen Et Al. - 2012 - Friedreich's Ataxia Causes Redistribution of Ironfidel muñozNoch keine Bewertungen
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceVon EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceBewertung: 4 von 5 Sternen4/5 (890)
- The Yellow House: A Memoir (2019 National Book Award Winner)Von EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Bewertung: 4 von 5 Sternen4/5 (98)
- The Little Book of Hygge: Danish Secrets to Happy LivingVon EverandThe Little Book of Hygge: Danish Secrets to Happy LivingBewertung: 3.5 von 5 Sternen3.5/5 (399)
- On Fire: The (Burning) Case for a Green New DealVon EverandOn Fire: The (Burning) Case for a Green New DealBewertung: 4 von 5 Sternen4/5 (73)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeVon EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeBewertung: 4 von 5 Sternen4/5 (5794)
- Never Split the Difference: Negotiating As If Your Life Depended On ItVon EverandNever Split the Difference: Negotiating As If Your Life Depended On ItBewertung: 4.5 von 5 Sternen4.5/5 (838)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureVon EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureBewertung: 4.5 von 5 Sternen4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryVon EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryBewertung: 3.5 von 5 Sternen3.5/5 (231)
- The Emperor of All Maladies: A Biography of CancerVon EverandThe Emperor of All Maladies: A Biography of CancerBewertung: 4.5 von 5 Sternen4.5/5 (271)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreVon EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreBewertung: 4 von 5 Sternen4/5 (1090)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyVon EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyBewertung: 3.5 von 5 Sternen3.5/5 (2219)
- Team of Rivals: The Political Genius of Abraham LincolnVon EverandTeam of Rivals: The Political Genius of Abraham LincolnBewertung: 4.5 von 5 Sternen4.5/5 (234)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersVon EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersBewertung: 4.5 von 5 Sternen4.5/5 (344)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaVon EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaBewertung: 4.5 von 5 Sternen4.5/5 (265)
- The Unwinding: An Inner History of the New AmericaVon EverandThe Unwinding: An Inner History of the New AmericaBewertung: 4 von 5 Sternen4/5 (45)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Von EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Bewertung: 4.5 von 5 Sternen4.5/5 (119)
- Her Body and Other Parties: StoriesVon EverandHer Body and Other Parties: StoriesBewertung: 4 von 5 Sternen4/5 (821)
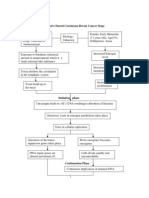
- Pathophysiology Invasive Ductal Carcinoma Breast Cancer StagesDokument3 SeitenPathophysiology Invasive Ductal Carcinoma Breast Cancer StagesAngelique Ramos Pascua100% (1)
- Sydney Telischak ResumeDokument2 SeitenSydney Telischak Resumeapi-546817891Noch keine Bewertungen
- List of Nanda Nursing Diagnosis 2012Dokument6 SeitenList of Nanda Nursing Diagnosis 2012Amit MartinNoch keine Bewertungen
- Test Bank of Neurocognitive DisordersDokument27 SeitenTest Bank of Neurocognitive DisordersmNoch keine Bewertungen
- 3D CD Technique Completes Dentures in 3 Days for ElderlyDokument5 Seiten3D CD Technique Completes Dentures in 3 Days for ElderlyMaulida Dara HarjantiNoch keine Bewertungen
- Chemotherapy ToxicityDokument37 SeitenChemotherapy ToxicityCarlos Eduardo Cadu100% (1)
- Froelich Syndrome and HomoeopathyDokument13 SeitenFroelich Syndrome and HomoeopathyDr. Rajneesh Kumar Sharma MD Hom100% (1)
- Urinary Tract Disorders, PowerpointDokument63 SeitenUrinary Tract Disorders, Powerpointmutia mutia100% (4)
- Catalogo AlergiaDokument60 SeitenCatalogo AlergiaMoreira SuNoch keine Bewertungen
- ProjectDokument64 SeitenProjectPradip DindaNoch keine Bewertungen
- Coxsackievirus: Presented By: LKCDokument18 SeitenCoxsackievirus: Presented By: LKCLeang KarichakNoch keine Bewertungen
- Sign Up For Our Enews: Chronic Wasting DiseaseDokument4 SeitenSign Up For Our Enews: Chronic Wasting DiseasesakuraleeshaoranNoch keine Bewertungen
- Analisis Kebutuhan Sumber Daya Manusia Terhadap Beban Kerja Di Bagian Rekam Medis Menggunakan Metode (Work Load Indicator Tahun 2019Dokument13 SeitenAnalisis Kebutuhan Sumber Daya Manusia Terhadap Beban Kerja Di Bagian Rekam Medis Menggunakan Metode (Work Load Indicator Tahun 2019roraNoch keine Bewertungen
- Herpes Virus 1 & Herpes Virus 2Dokument80 SeitenHerpes Virus 1 & Herpes Virus 2rehanaNoch keine Bewertungen
- Drugs To Watch With WARFARINDokument3 SeitenDrugs To Watch With WARFARINRajendra RaiNoch keine Bewertungen
- Saudi Internship GuideDokument47 SeitenSaudi Internship GuideRizky KykyNoch keine Bewertungen
- Circulating SerotoninDokument17 SeitenCirculating Serotoninnihilx27374Noch keine Bewertungen
- Moh Dha MaterialsDokument92 SeitenMoh Dha Materialswafaa al tawil100% (2)
- Dialysis BrochureDokument13 SeitenDialysis BrochureRaffy FlorentinoNoch keine Bewertungen
- Careers in Medicine CV Sample #4Dokument3 SeitenCareers in Medicine CV Sample #4huyenthanh1807Noch keine Bewertungen
- GGT InsertDokument2 SeitenGGT InsertsharmashyamsinghNoch keine Bewertungen
- Scopus Indexed Journal PDFDokument330 SeitenScopus Indexed Journal PDFDharmendra PrajapatiNoch keine Bewertungen
- Hot Sitz Bath ReliefDokument10 SeitenHot Sitz Bath ReliefJohn Aladin ArcetaNoch keine Bewertungen
- G1a115016 - Tesa SeptiariDokument12 SeitenG1a115016 - Tesa SeptiariTesa SeptiariNoch keine Bewertungen
- LECTURE 4 Introduction To MicrobiologyDokument36 SeitenLECTURE 4 Introduction To MicrobiologyArcee Feb Dela PazNoch keine Bewertungen
- Kertas Intervensi-EvaluasiDokument7 SeitenKertas Intervensi-EvaluasiNURUL AWALIAHNoch keine Bewertungen
- OMCDokument37 SeitenOMCyurie_ameliaNoch keine Bewertungen
- 1926, August 12 New JerseyDokument2 Seiten1926, August 12 New JerseyKiara Catelyn RetretaNoch keine Bewertungen
- What Is AmenorrheaDokument5 SeitenWhat Is AmenorrheaYhoyho Akhilun Dewa MimpiNoch keine Bewertungen
- Anesthesiologists and AddictionDokument8 SeitenAnesthesiologists and AddictionCalli DerosierNoch keine Bewertungen