Das könnte Ihnen auch gefallen
- Equipment Qualification in the Pharmaceutical IndustryVon EverandEquipment Qualification in the Pharmaceutical IndustryBewertung: 3.5 von 5 Sternen3.5/5 (3)
- Principles of Parenteral Solution Validation: A Practical Lifecycle ApproachVon EverandPrinciples of Parenteral Solution Validation: A Practical Lifecycle ApproachIgor GorskyBewertung: 5 von 5 Sternen5/5 (2)
- Practical Approaches to Method Validation and Essential Instrument QualificationVon EverandPractical Approaches to Method Validation and Essential Instrument QualificationNoch keine Bewertungen
- ECA Task Force CCS Guideline Vers2Dokument53 SeitenECA Task Force CCS Guideline Vers2elisabetta ghilardi100% (2)
- Risk Management in Sterile EnvironmentsDokument30 SeitenRisk Management in Sterile EnvironmentsTim Sandle100% (4)
- Pda TR 54Dokument79 SeitenPda TR 54Claudia Marcela Gómez100% (1)
- Biocontamination Control for Pharmaceuticals and HealthcareVon EverandBiocontamination Control for Pharmaceuticals and HealthcareBewertung: 5 von 5 Sternen5/5 (1)
- PDA TR 80 《制药实验室数据完整性管理体系》中英文对照版Dokument98 SeitenPDA TR 80 《制药实验室数据完整性管理体系》中英文对照版于天一100% (1)
- Cleanroom Technology: Fundamentals of Design, Testing and OperationVon EverandCleanroom Technology: Fundamentals of Design, Testing and OperationNoch keine Bewertungen
- TR 35 A Proposed Training Model For The Microbiological Function in The Pharmaceutical Industry 2001Dokument32 SeitenTR 35 A Proposed Training Model For The Microbiological Function in The Pharmaceutical Industry 2001Sheetal Agree100% (2)
- PDA - Journal - PST - Volume 75 Number 5 Sep OCt 2021Dokument74 SeitenPDA - Journal - PST - Volume 75 Number 5 Sep OCt 2021anh tuan ngo100% (1)
- Media Fills and Environment Atl Monitoring 26 July 2011 Presentation OneDokument18 SeitenMedia Fills and Environment Atl Monitoring 26 July 2011 Presentation OneRiccardo TorelliNoch keine Bewertungen
- TocDokument7 SeitenTocIlayaraja Boopathy0% (1)
- Burkholderia Cepacia Complex: Characteristics, Products Risks and Testing RequirementsDokument17 SeitenBurkholderia Cepacia Complex: Characteristics, Products Risks and Testing RequirementsTim Sandle100% (1)
- ECA Guidelines For The Evaluation and Investigation of Microbiological DeviationsDokument33 SeitenECA Guidelines For The Evaluation and Investigation of Microbiological Deviationsmmmmm100% (5)
- Production of Plasma Proteins for Therapeutic UseVon EverandProduction of Plasma Proteins for Therapeutic UseBewertung: 3 von 5 Sternen3/5 (5)
- Media Fill ChecklistDokument11 SeitenMedia Fill ChecklistSilke Igemann100% (1)
- Clenaing ValidationDokument31 SeitenClenaing Validationagarciah15891100% (2)
- Risk Assessment and Management For Healthcare Manufacturing: Practical Tips and Case StudiesDokument14 SeitenRisk Assessment and Management For Healthcare Manufacturing: Practical Tips and Case StudiesTim SandleNoch keine Bewertungen
- Risk Management Applications in Pharmaceutical and Biopharmaceutical ManufacturingVon EverandRisk Management Applications in Pharmaceutical and Biopharmaceutical ManufacturingHamid MollahNoch keine Bewertungen
- Env Monitoring Cleanrooms Final PDFDokument37 SeitenEnv Monitoring Cleanrooms Final PDFnsk79in@gmail.comNoch keine Bewertungen
- Disinfectants in Pharmaceutical Industry Tim SandleDokument8 SeitenDisinfectants in Pharmaceutical Industry Tim Sandleshwampa100% (1)
- PDA TR 65 Technology Transfer技术转移-中英对照GELGEDokument85 SeitenPDA TR 65 Technology Transfer技术转移-中英对照GELGECelven Shr100% (1)
- PDART59 12 Statistical MethodsDokument74 SeitenPDART59 12 Statistical MethodsJorge Martinez Quezada100% (2)
- Pdajpst 2015 01079Dokument21 SeitenPdajpst 2015 01079ErikaNoch keine Bewertungen
- Case Study: Risk-Based Approach To Containment and Control For Potent/ Hazardous CompoundsDokument8 SeitenCase Study: Risk-Based Approach To Containment and Control For Potent/ Hazardous CompoundsXCASTERAD100% (1)
- Cleaning Validation Master Plan PDFDokument9 SeitenCleaning Validation Master Plan PDFBREWSKINoch keine Bewertungen
- Media Fill FDA 483 ObservationsDokument22 SeitenMedia Fill FDA 483 Observationsvijayns_250355172Noch keine Bewertungen
- Pda Technical Report 48 Moist Heat Sterilizer SystemsDokument70 SeitenPda Technical Report 48 Moist Heat Sterilizer SystemsDuc100% (2)
- Good Manufacturing Practices (GMP) Modules for Pharmaceutical ProductsVon EverandGood Manufacturing Practices (GMP) Modules for Pharmaceutical ProductsNoch keine Bewertungen
- Media Fill Tech Re PDFDokument21 SeitenMedia Fill Tech Re PDFnetelsrt1298100% (1)
- Establishing A Complete Set of Target, Alert, and Action Limits For Microbial Counts in Purified WaterDokument8 SeitenEstablishing A Complete Set of Target, Alert, and Action Limits For Microbial Counts in Purified WaterSagi Nguyen100% (2)
- The Media Fill Approach An UpdateDokument3 SeitenThe Media Fill Approach An UpdateajmalnasirNoch keine Bewertungen
- PDA Journal SeptemberDokument19 SeitenPDA Journal SeptemberZia100% (2)
- Gamma Radiation SterilizationDokument2 SeitenGamma Radiation Sterilizationananth67% (3)
- Pharmaceutical Quality by Design: A Practical ApproachVon EverandPharmaceutical Quality by Design: A Practical ApproachWalkiria S. SchlindweinNoch keine Bewertungen
- PHSS Control Strategy White PaperDokument13 SeitenPHSS Control Strategy White PaperAkuWilliams100% (1)
- 1 Ispe Pat Cop Dach Awareness Doc Final v1.0Dokument67 Seiten1 Ispe Pat Cop Dach Awareness Doc Final v1.0lutfhi zarkasyi100% (1)
- 04 ISPEs Guides and How They Apply To Cleaning and Cleaning Validation by Stephanie WilkinsDokument33 Seiten04 ISPEs Guides and How They Apply To Cleaning and Cleaning Validation by Stephanie Wilkinsmjamil0995100% (1)
- IVT Network - A Risk Matrix Approach For Media Simulation Trials - 2014-04-30Dokument11 SeitenIVT Network - A Risk Matrix Approach For Media Simulation Trials - 2014-04-30vijayns_250355172Noch keine Bewertungen
- IVT Network - Microbiological Assessment of Compressed Gases in Pharmaceutical Facilities - 2015-08-17Dokument7 SeitenIVT Network - Microbiological Assessment of Compressed Gases in Pharmaceutical Facilities - 2015-08-17Youstina PhillipeNoch keine Bewertungen
- The Manufacture of Sterile Pharmaceuticals and Liquid Medical Devices Using Blow-Fill-Seal Technology: Points to ConsiderVon EverandThe Manufacture of Sterile Pharmaceuticals and Liquid Medical Devices Using Blow-Fill-Seal Technology: Points to ConsiderNoch keine Bewertungen
- Risk Assessment Considerations For Installation of A New AutoclaveDokument29 SeitenRisk Assessment Considerations For Installation of A New AutoclaveDoan Chi ThienNoch keine Bewertungen
- Microbiological Enviromental MonitoringDokument34 SeitenMicrobiological Enviromental MonitoringAna Dulce100% (1)
- PDA Technical Report 22 - SPADokument50 SeitenPDA Technical Report 22 - SPAErika100% (2)
- ISPE Cleaningvalidation PDFDokument42 SeitenISPE Cleaningvalidation PDFAjay KumarNoch keine Bewertungen
- Cleaning Validation Guidelines - A Complete ListDokument11 SeitenCleaning Validation Guidelines - A Complete ListSanjay SharmaNoch keine Bewertungen
- PDA Journal Volume 75 Issue 2.2021Dokument97 SeitenPDA Journal Volume 75 Issue 2.2021anh tuan ngo100% (1)
- Cleaning Validation Report Dec2018 PDFDokument21 SeitenCleaning Validation Report Dec2018 PDFPrashansa ShresthaNoch keine Bewertungen
- PDA TR Nº40 Sterilizing Filtration of Gases PDFDokument45 SeitenPDA TR Nº40 Sterilizing Filtration of Gases PDFehsan050628Noch keine Bewertungen
- Regulatory Aspects of Pharmaceutical Quality System: Brief IntroductionVon EverandRegulatory Aspects of Pharmaceutical Quality System: Brief IntroductionNoch keine Bewertungen
- cGMP Current Good Manufacturing Practices for PharmaceuticalsVon EverandcGMP Current Good Manufacturing Practices for PharmaceuticalsBewertung: 1 von 5 Sternen1/5 (2)
- ICH Quality Guidelines: An Implementation GuideVon EverandICH Quality Guidelines: An Implementation GuideAndrew TeasdaleNoch keine Bewertungen
- Principles and Practices of Contamination Control and CleanroomsVon EverandPrinciples and Practices of Contamination Control and CleanroomsNoch keine Bewertungen
- Batch DispositionDokument21 SeitenBatch DispositionNUPharma Consular100% (1)
- Draft Annex 1 Contamination Control Strategy For Barrier Systems Like IsolatorsDokument33 SeitenDraft Annex 1 Contamination Control Strategy For Barrier Systems Like IsolatorsWiktor Pszczółkowski100% (1)
- Microsoft PowerPoint - 20.10 11.30 - Sterile Medicinal ProductsDokument35 SeitenMicrosoft PowerPoint - 20.10 11.30 - Sterile Medicinal Productsdavincicode888Noch keine Bewertungen
- IVT Network - API Pharmaceutical Water Systems Part I - Water System Design - 2014-06-13Dokument5 SeitenIVT Network - API Pharmaceutical Water Systems Part I - Water System Design - 2014-06-13davincicode888Noch keine Bewertungen
- 1.check The Appropriate Quantities of Sterile Ceftriaxone Sodium 2.preparation and Sterilization/depyrogenation of Glass Vials, EquipmentsDokument1 Seite1.check The Appropriate Quantities of Sterile Ceftriaxone Sodium 2.preparation and Sterilization/depyrogenation of Glass Vials, Equipmentsdavincicode888Noch keine Bewertungen
- IVTJVT0512 Aseptic FinalDokument7 SeitenIVTJVT0512 Aseptic Finaldavincicode888Noch keine Bewertungen
- IVTJVT0512 Aseptic FinalDokument7 SeitenIVTJVT0512 Aseptic Finaldavincicode888Noch keine Bewertungen
- The Environmental Monitoring Program in A GMP Environment: Microbiology TopicsDokument9 SeitenThe Environmental Monitoring Program in A GMP Environment: Microbiology Topicsdavincicode888Noch keine Bewertungen
- P P C O N ' D W: Osed Ithout RescriptionDokument64 SeitenP P C O N ' D W: Osed Ithout Rescriptiondavincicode888Noch keine Bewertungen
- ISO 9000:2015 Quality Management: Systems - Fundamentals and VocabularyDokument6 SeitenISO 9000:2015 Quality Management: Systems - Fundamentals and Vocabularydavincicode888Noch keine Bewertungen
- Hot Topics in The Visual Inspection of Injectable ProductsDokument46 SeitenHot Topics in The Visual Inspection of Injectable Productsdavincicode888Noch keine Bewertungen
- Environmental Monitoring Management in Pharmaceutical Facilities To Comply With Pic GMP Requirements Sapphire108Dokument36 SeitenEnvironmental Monitoring Management in Pharmaceutical Facilities To Comply With Pic GMP Requirements Sapphire108davincicode888100% (1)
- Mark PDFDokument42 SeitenMark PDFdavincicode888Noch keine Bewertungen
- WhitePaper Invensys LifeSciencesIndustrySolutionBlueprintForEMS-IsPE 08-11Dokument15 SeitenWhitePaper Invensys LifeSciencesIndustrySolutionBlueprintForEMS-IsPE 08-11davincicode888Noch keine Bewertungen
- Container and Closure System IntegrityDokument9 SeitenContainer and Closure System Integritydavincicode888Noch keine Bewertungen
- Pharm Life SciencesDokument93 SeitenPharm Life Sciencesdavincicode888Noch keine Bewertungen
- Thai FDA 49 Pics Pa - SuchartDokument23 SeitenThai FDA 49 Pics Pa - Suchartdavincicode888Noch keine Bewertungen
- On Demand Webinar Slides Ensuring USP Compliance enDokument66 SeitenOn Demand Webinar Slides Ensuring USP Compliance endavincicode888Noch keine Bewertungen
- BPMDokument3 SeitenBPMarunsawaiyanNoch keine Bewertungen
- Reading 1 - Science - BIOL105 - Disease - ResponseDokument9 SeitenReading 1 - Science - BIOL105 - Disease - ResponsemykrohNoch keine Bewertungen
- Unitec Research Committee Final Report: Researcher: Project Title: Project Code: Date of ReportDokument6 SeitenUnitec Research Committee Final Report: Researcher: Project Title: Project Code: Date of ReportRiya JosephNoch keine Bewertungen
- EDUC5271 Week 3 Written Assignment Unit 3Dokument7 SeitenEDUC5271 Week 3 Written Assignment Unit 3Meleisa GordonNoch keine Bewertungen
- Data Communication and Computer Networks (EIE418) : Prof. E. Adetiba (PH.D, R.Engr. (COREN) )Dokument52 SeitenData Communication and Computer Networks (EIE418) : Prof. E. Adetiba (PH.D, R.Engr. (COREN) )John DavidNoch keine Bewertungen
- Application For Petitioned Class: College of Engineering and ArchitectureDokument2 SeitenApplication For Petitioned Class: College of Engineering and ArchitectureJohn A. CenizaNoch keine Bewertungen
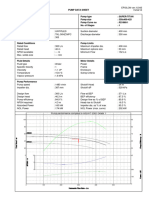
- TKL Pump - Data - SheetDokument1 SeiteTKL Pump - Data - Sheetธนาชัย เต็งจิรธนาภาNoch keine Bewertungen
- 20110715122354-B SC - M SC - H S - Mathematics PDFDokument75 Seiten20110715122354-B SC - M SC - H S - Mathematics PDFn0% (1)
- Program in Less Than 24 MonthsDokument2 SeitenProgram in Less Than 24 MonthsHermi BurquesNoch keine Bewertungen
- Yolo v4Dokument42 SeitenYolo v4Deepak SaxenaNoch keine Bewertungen
- 7.proceeding Snib-Eng Dept, Pnp-IndonesiaDokument11 Seiten7.proceeding Snib-Eng Dept, Pnp-IndonesiamissfifitNoch keine Bewertungen
- Using Fonts Installed in Local Texlive - TeX - LaTeX Stack ExchangeDokument8 SeitenUsing Fonts Installed in Local Texlive - TeX - LaTeX Stack ExchangeFuncionario CepaaNoch keine Bewertungen
- Ensci 200 Fall 2021 Writting RubricDokument1 SeiteEnsci 200 Fall 2021 Writting RubricAsheNoch keine Bewertungen
- DISS TOS Q3 TESTDokument3 SeitenDISS TOS Q3 TESTMichelle GariandoNoch keine Bewertungen
- Social Construction of DisasterDokument385 SeitenSocial Construction of DisasterPratik Kumar BandyopadhyayNoch keine Bewertungen
- Lec 5 (Welded Joint)Dokument38 SeitenLec 5 (Welded Joint)Ahmed HassanNoch keine Bewertungen
- Who Am I MemoDokument2 SeitenWho Am I Memoapi-652685391Noch keine Bewertungen
- 21st Bomber Command Tactical Mission Report 64, 65, OcrDokument57 Seiten21st Bomber Command Tactical Mission Report 64, 65, OcrJapanAirRaidsNoch keine Bewertungen
- Analysis of Transfer Lines: UNIT-3 Automation Manufracturing PvpsitDokument14 SeitenAnalysis of Transfer Lines: UNIT-3 Automation Manufracturing PvpsitSravanth KondetiNoch keine Bewertungen
- 4 A Study of Encryption AlgorithmsDokument9 Seiten4 A Study of Encryption AlgorithmsVivekNoch keine Bewertungen
- Lorma Scope of WorkDokument2 SeitenLorma Scope of WorkJb TiscubNoch keine Bewertungen
- Enerizons Presentation 2018Dokument49 SeitenEnerizons Presentation 2018Hussien El SheikhNoch keine Bewertungen
- STPM 954 Math T Coursework 2013 Sem 2Dokument8 SeitenSTPM 954 Math T Coursework 2013 Sem 2vtdvkkjbf100% (2)
- Candy System Functionalities ListDokument4 SeitenCandy System Functionalities ListPauloKupesaNoch keine Bewertungen
- Growth Mindset Intermediate Lesson Plans The Ned ShowDokument16 SeitenGrowth Mindset Intermediate Lesson Plans The Ned ShowGôis Madson100% (2)
- SY2021 2022 2ndsemesterDokument2 SeitenSY2021 2022 2ndsemesterWilfredo III DayagdagNoch keine Bewertungen
- Improved Mini-Silo For Studying Fermentation of Silage in Laboratory ConditionsDokument8 SeitenImproved Mini-Silo For Studying Fermentation of Silage in Laboratory ConditionsCk_psihNoch keine Bewertungen
- Aster 30 Sampt PDFDokument2 SeitenAster 30 Sampt PDFmig29Noch keine Bewertungen
- Data 6005ADokument1 SeiteData 6005AErick De La RoSaNoch keine Bewertungen
- Distributed Caching Algorithms For Content Distribution NetworksDokument22 SeitenDistributed Caching Algorithms For Content Distribution NetworksgodgivenhomesNoch keine Bewertungen