Das könnte Ihnen auch gefallen
- Cleaning Validation SOP Novartis PDFDokument15 SeitenCleaning Validation SOP Novartis PDFFaisal Abbas100% (2)
- TEM-280 Packaging Validation Protocol Template SampleDokument6 SeitenTEM-280 Packaging Validation Protocol Template SampleAnonymous BcT42WLn50% (2)
- Validating Cleaning Processes Used During The Manufacture of Medical DevicesDokument16 SeitenValidating Cleaning Processes Used During The Manufacture of Medical DevicesFabricio Amorim100% (2)
- Validating Cleaning Processes Used During The Manufacture of Medical DevicesDokument16 SeitenValidating Cleaning Processes Used During The Manufacture of Medical DevicesFabricio Amorim100% (2)
- Hold Time Study of Cleaned EquipmentsDokument3 SeitenHold Time Study of Cleaned EquipmentsShubam Sharma60% (5)
- Validation Plan TemplateDokument23 SeitenValidation Plan Templatedes50% (2)
- Retaining Wall DetailDokument9 SeitenRetaining Wall DetailRonald KahoraNoch keine Bewertungen
- CIQA Validation Master Plan Sample TemplateDokument4 SeitenCIQA Validation Master Plan Sample TemplateSatyam Gupta100% (1)
- Bulk Holding Time Study ReportDokument8 SeitenBulk Holding Time Study ReportFaress RabiNoch keine Bewertungen
- CCS Guide Attachment3 v2Dokument34 SeitenCCS Guide Attachment3 v2elisabetta ghilardi100% (1)
- 023-SOP For Conducting Hold Time StudyDokument3 Seiten023-SOP For Conducting Hold Time StudyAshok Lenka100% (2)
- Disinfectant Efficacy ValidationDokument12 SeitenDisinfectant Efficacy Validationhbhatt8890% (10)
- Temperature Mapping Study Protocol Procedure PDFDokument15 SeitenTemperature Mapping Study Protocol Procedure PDFJhonnnnnnNoch keine Bewertungen
- Cleaning Validation Guidelines - A Complete ListDokument11 SeitenCleaning Validation Guidelines - A Complete ListSanjay SharmaNoch keine Bewertungen
- Helix Pharma (Private) Limited: Validation ProtocolDokument4 SeitenHelix Pharma (Private) Limited: Validation Protocolziauddin bukhari0% (2)
- Validation ProtocolDokument9 SeitenValidation ProtocolVikram ChhabraNoch keine Bewertungen
- SOP - Clothing Requirements Inside The FactoryDokument3 SeitenSOP - Clothing Requirements Inside The Factorypiyusharora1964Noch keine Bewertungen
- 30 ML Moulded Vial Filling OQDokument15 Seiten30 ML Moulded Vial Filling OQSubhash NaiduNoch keine Bewertungen
- Performance Qualification Protocol For Water For Injection (WFI) System - Pharmaceutical GuidelinesDokument3 SeitenPerformance Qualification Protocol For Water For Injection (WFI) System - Pharmaceutical GuidelinesJayesh PatidarNoch keine Bewertungen
- Validation of Coating Equipment (Ketik Ulang)Dokument6 SeitenValidation of Coating Equipment (Ketik Ulang)Dedhieaja0% (1)
- Cotton Web ReportDokument30 SeitenCotton Web ReportHumaRiaz33% (3)
- Pharmaceutical Industry Documents: 90 Pharmaceutical Quality Assurance Interview Questions & AnswersVon EverandPharmaceutical Industry Documents: 90 Pharmaceutical Quality Assurance Interview Questions & AnswersNoch keine Bewertungen
- Establishing A CGMP Laboratory Audit System: A Practical GuideVon EverandEstablishing A CGMP Laboratory Audit System: A Practical GuideNoch keine Bewertungen
- Cleaning Validation Protocol TEMPLATEDokument9 SeitenCleaning Validation Protocol TEMPLATEnatavceNoch keine Bewertungen
- Equipment Logbook 2 2Dokument7 SeitenEquipment Logbook 2 2Belazouz BoualemNoch keine Bewertungen
- Juvia Protocol 50-1000Dokument16 SeitenJuvia Protocol 50-1000ziauddin bukhariNoch keine Bewertungen
- Cleaning Validation Sample ProtocolDokument7 SeitenCleaning Validation Sample ProtocolArieTamaNoch keine Bewertungen
- Annual Product Review Developing An SOPDokument26 SeitenAnnual Product Review Developing An SOPanants2567% (3)
- Title: Cleaning Validation Report For - ToDokument41 SeitenTitle: Cleaning Validation Report For - TocpkakopeNoch keine Bewertungen
- Validation Protocol For Efficacy of Chemical DisinfectantsDokument8 SeitenValidation Protocol For Efficacy of Chemical DisinfectantsMohamed KamalNoch keine Bewertungen
- Validation Protocol CG TADokument30 SeitenValidation Protocol CG TACarlo Duran100% (1)
- Master Cleaning Validation PlanDokument25 SeitenMaster Cleaning Validation PlanWidya Lukitasari100% (1)
- 8.3 Operation Qualification Protocol For Dispensing BoothDokument4 Seiten8.3 Operation Qualification Protocol For Dispensing BoothTanveer Ahmed Quadri100% (1)
- TEMPLATE FOR PROCESS VALIDATION PROTOCOL - Pharmaceutical GuidanceDokument6 SeitenTEMPLATE FOR PROCESS VALIDATION PROTOCOL - Pharmaceutical GuidancePackaging Development BernofarmNoch keine Bewertungen
- Process Validation Sample Protocol - PharmaguidelineDokument3 SeitenProcess Validation Sample Protocol - PharmaguidelineD Tech Dental Technologies100% (1)
- Site VMPDokument34 SeitenSite VMPJonatan Dominguez PerezNoch keine Bewertungen
- Parenteral Process Validation 1Dokument30 SeitenParenteral Process Validation 1BALU LTD BALU PHARMACEUTICALNoch keine Bewertungen
- Installation Qualification Protocol For Walk - in Incubator System Name: Walk - in Incubator Document No.: NECPL/IQ/11220I/1 Page No.: 1 of 25Dokument25 SeitenInstallation Qualification Protocol For Walk - in Incubator System Name: Walk - in Incubator Document No.: NECPL/IQ/11220I/1 Page No.: 1 of 25vipin100% (1)
- Process Validation of LiquidDokument24 SeitenProcess Validation of LiquidAshutosh Shukla100% (2)
- Hold Time Study 1Dokument3 SeitenHold Time Study 1aboemadaldeenNoch keine Bewertungen
- Hold Time Study For Cleaned FBD BagsDokument6 SeitenHold Time Study For Cleaned FBD BagsDevendra Dwivedi100% (1)
- PVPRDokument47 SeitenPVPRBRIJENDRA KUMAR SINGH100% (2)
- Texwipe PDA Cleaning and Cleaning Validation Chapter19Dokument26 SeitenTexwipe PDA Cleaning and Cleaning Validation Chapter19davincicode888100% (1)
- Validation VialWasher OQ NIHDokument30 SeitenValidation VialWasher OQ NIHcongacon3aNoch keine Bewertungen
- N Hold Time StudyDokument16 SeitenN Hold Time StudyziadddNoch keine Bewertungen
- Report Approval Sheet: Modi Sugar Mills, ModinagarDokument7 SeitenReport Approval Sheet: Modi Sugar Mills, Modinagar9889187549100% (2)
- SOP For Conduct Temperature Mapping in StoresDokument3 SeitenSOP For Conduct Temperature Mapping in StoresSolomon Gamanuel0% (1)
- 9 C Validation Protocol TABLETDokument20 Seiten9 C Validation Protocol TABLETMohammed ZubairNoch keine Bewertungen
- Ointment Process ValidationDokument25 SeitenOintment Process ValidationTrinh Huy CongNoch keine Bewertungen
- Batch DispositionDokument21 SeitenBatch DispositionNUPharma Consular100% (1)
- Cleaning Validation ProtocolDokument9 SeitenCleaning Validation Protocolyash143565100% (2)
- Annex4-TRS992 Hold Time Study GuidelineDokument8 SeitenAnnex4-TRS992 Hold Time Study Guidelinensk79in@gmail.com100% (1)
- Cleaning Validation ProtocolDokument16 SeitenCleaning Validation ProtocolRutvik59100% (3)
- Operational Qualification For Compressed Air System.Dokument11 SeitenOperational Qualification For Compressed Air System.BREWSKI50% (2)
- F03qa038-00 VMPDokument24 SeitenF03qa038-00 VMPMeet Vermaa100% (1)
- URS Lab Scale Counter Pressure Autoclave (Rev 1b)Dokument12 SeitenURS Lab Scale Counter Pressure Autoclave (Rev 1b)puneetogupta100% (2)
- Validation Master Plan A Complete Guide - 2020 EditionVon EverandValidation Master Plan A Complete Guide - 2020 EditionNoch keine Bewertungen
- Biocontamination Control for Pharmaceuticals and HealthcareVon EverandBiocontamination Control for Pharmaceuticals and HealthcareBewertung: 5 von 5 Sternen5/5 (1)
- Regulatory Aspects of Pharmaceutical Quality System: Brief IntroductionVon EverandRegulatory Aspects of Pharmaceutical Quality System: Brief IntroductionNoch keine Bewertungen
- Good Distribution Practice A Complete Guide - 2021 EditionVon EverandGood Distribution Practice A Complete Guide - 2021 EditionNoch keine Bewertungen
- Production of Plasma Proteins for Therapeutic UseVon EverandProduction of Plasma Proteins for Therapeutic UseBewertung: 3 von 5 Sternen3/5 (5)
- Cleanroom Technology: Fundamentals of Design, Testing and OperationVon EverandCleanroom Technology: Fundamentals of Design, Testing and OperationNoch keine Bewertungen
- OQ & PQ Medical DeviceDokument13 SeitenOQ & PQ Medical DeviceBREWSKI100% (1)
- Process Validation and Revalidation in Medical Device ProductionDokument7 SeitenProcess Validation and Revalidation in Medical Device ProductionBREWSKINoch keine Bewertungen
- Process Validation and Revalidation in Medical Device ProductionDokument7 SeitenProcess Validation and Revalidation in Medical Device ProductionBREWSKINoch keine Bewertungen
- Ipf2018 Presentation21Dokument192 SeitenIpf2018 Presentation21Vinoth JothiNoch keine Bewertungen
- Risk MGT Template IT Pharma Validation EuropeDokument11 SeitenRisk MGT Template IT Pharma Validation EuropeBREWSKINoch keine Bewertungen
- SER Equirements Emplate OR A Ully Utomatic Pindle Apper: User Requirements Specification CapperDokument26 SeitenSER Equirements Emplate OR A Ully Utomatic Pindle Apper: User Requirements Specification CapperBREWSKINoch keine Bewertungen
- Operating Instructions Coding and Inspection Unit TQS-LM (Line Manager)Dokument94 SeitenOperating Instructions Coding and Inspection Unit TQS-LM (Line Manager)BREWSKINoch keine Bewertungen
- U R T Foraw R L F: Ser Equirements Emplate Ide Ange Iquid IllerDokument23 SeitenU R T Foraw R L F: Ser Equirements Emplate Ide Ange Iquid IllerBREWSKINoch keine Bewertungen
- URS Contents: Blank TemplateDokument11 SeitenURS Contents: Blank TemplateBREWSKI100% (1)
- Cleaning Validation StandardDokument14 SeitenCleaning Validation StandardBREWSKINoch keine Bewertungen
- Process Validation and Revalidation in Medical Device ProductionDokument7 SeitenProcess Validation and Revalidation in Medical Device ProductionBREWSKINoch keine Bewertungen
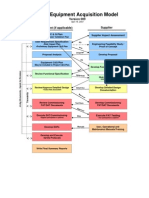
- AcquisitionModelVer9 PDFDokument5 SeitenAcquisitionModelVer9 PDFantonygamalpharma100% (1)
- Sealing Process Validation Guideline - enDokument14 SeitenSealing Process Validation Guideline - enyadu100% (1)
- Risk MGT Template IT Pharma Validation EuropeDokument11 SeitenRisk MGT Template IT Pharma Validation EuropeBREWSKINoch keine Bewertungen
- Urs Template ContentsDokument8 SeitenUrs Template ContentsipatoffNoch keine Bewertungen
- NT - Akivision A CFRDokument60 SeitenNT - Akivision A CFRBREWSKINoch keine Bewertungen
- JETT Document Status Tracking WorksheetDokument6 SeitenJETT Document Status Tracking WorksheetBREWSKINoch keine Bewertungen
- Jett Urs P P: Reparation RocessDokument7 SeitenJett Urs P P: Reparation RocessBREWSKINoch keine Bewertungen
- ISPE Statistical MethodDokument137 SeitenISPE Statistical MethodDyah W K Antiek100% (1)
- NT - Akivision A CFRDokument60 SeitenNT - Akivision A CFRBREWSKINoch keine Bewertungen
- Change Control Risk AssessmentDokument1 SeiteChange Control Risk AssessmentBREWSKINoch keine Bewertungen
- Urs Template ContentsDokument8 SeitenUrs Template ContentsipatoffNoch keine Bewertungen
- NT - Akivision A CFRDokument60 SeitenNT - Akivision A CFRBREWSKINoch keine Bewertungen
- CPV Case Study Interactive VersionDokument52 SeitenCPV Case Study Interactive VersionBREWSKINoch keine Bewertungen
- Change Control Risk AssessmentDokument1 SeiteChange Control Risk AssessmentBREWSKINoch keine Bewertungen
- Texwipe PDA Cleaning and Cleaning Validation Chapter19Dokument26 SeitenTexwipe PDA Cleaning and Cleaning Validation Chapter19davincicode888100% (1)
- Whitepaper Serialisation 150127Dokument3 SeitenWhitepaper Serialisation 150127BREWSKINoch keine Bewertungen
- CRBWhitepaperSerialization eDokument8 SeitenCRBWhitepaperSerialization eBREWSKINoch keine Bewertungen
- 20.104 Construction of SteelworkDokument52 Seiten20.104 Construction of SteelworkWilliam Chong100% (2)
- Practice Perf CompDokument28 SeitenPractice Perf CompsooguyNoch keine Bewertungen
- MaterialDokument7 SeitenMaterialrajkamal_eNoch keine Bewertungen
- BiomassDokument36 SeitenBiomassGábor MátyásiNoch keine Bewertungen
- CH 7Dokument43 SeitenCH 7Hossam AliNoch keine Bewertungen
- Larsen & Toubro Limited: Ecc DivisionDokument4 SeitenLarsen & Toubro Limited: Ecc Divisionmanu_gite100% (2)
- Reference ListDokument28 SeitenReference ListThirukkumaranBalasubramanianNoch keine Bewertungen
- Ointment Process ValidationDokument25 SeitenOintment Process ValidationTrinh Huy CongNoch keine Bewertungen
- How To Make A Tool Post GrinderDokument5 SeitenHow To Make A Tool Post Grinderozland9Noch keine Bewertungen
- Palm Kernel Oil Mill ProjectDokument8 SeitenPalm Kernel Oil Mill Projectsjr141071100% (2)
- Acc320ptiipii SP12Dokument3 SeitenAcc320ptiipii SP12Melanie KilpatrickNoch keine Bewertungen
- Homeless Serving Land Use Overnight Shelter Parcel DataDokument14 SeitenHomeless Serving Land Use Overnight Shelter Parcel DataAbhishekh GuptaNoch keine Bewertungen
- Ribbon BlenderDokument3 SeitenRibbon BlenderjasenterpriseNoch keine Bewertungen
- Asme Sec Ix PT QB Article Xiv - Brazing DataDokument24 SeitenAsme Sec Ix PT QB Article Xiv - Brazing Datasamitha505Noch keine Bewertungen
- Renderoc Plug: Uses SpecificationDokument2 SeitenRenderoc Plug: Uses SpecificationVenkata RaoNoch keine Bewertungen
- TPM JH PPT 01 JH AwarenessDokument28 SeitenTPM JH PPT 01 JH AwarenessLakshmanan Venkatesan100% (1)
- Unscrewing Demold With SlidesDokument19 SeitenUnscrewing Demold With SlidesGhazouNoch keine Bewertungen
- WI Quality AssuranceDokument3 SeitenWI Quality Assurancearifin rizalNoch keine Bewertungen
- Indian Apparel Industry-An OverviewDokument41 SeitenIndian Apparel Industry-An OverviewAnil BatraNoch keine Bewertungen
- ASTM-A-325-02 Standard Specification For Structural Bolts. Steel, Heat Treated, 120 - 105 Ksi Minimum Tensile Strength PDFDokument8 SeitenASTM-A-325-02 Standard Specification For Structural Bolts. Steel, Heat Treated, 120 - 105 Ksi Minimum Tensile Strength PDFFattahi KarimNoch keine Bewertungen
- Le Van Sy PHD Thesis PDFDokument205 SeitenLe Van Sy PHD Thesis PDFNhan LeNoch keine Bewertungen
- Sika Duoflex NS/SL: Two-Component, Polysulphide SealantDokument2 SeitenSika Duoflex NS/SL: Two-Component, Polysulphide SealantAbu Vulkanik Al-JawiyNoch keine Bewertungen
- 06 Krempel CelluloseBaseInsulationMaterials PDFDokument29 Seiten06 Krempel CelluloseBaseInsulationMaterials PDFreza515heiNoch keine Bewertungen
- ASTM A510 03-Astm-Standardspecivication-1 PDFDokument7 SeitenASTM A510 03-Astm-Standardspecivication-1 PDFNina LazuardiNoch keine Bewertungen
- A Study On Inventory Management Towards Organizational Performance of Manufacturing Company in MelakaDokument12 SeitenA Study On Inventory Management Towards Organizational Performance of Manufacturing Company in MelakaOsama MazharNoch keine Bewertungen
- Presentation 1Dokument28 SeitenPresentation 1ikamelyaastutiNoch keine Bewertungen
- Management Accounting Wilkerson Company CasestudyDokument3 SeitenManagement Accounting Wilkerson Company CasestudysamacsterNoch keine Bewertungen
- DFM P2Dokument6 SeitenDFM P2Cosmin Ionuț BlaneteNoch keine Bewertungen