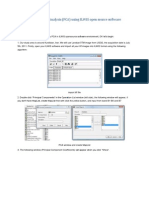
Das könnte Ihnen auch gefallen
- Chimera X TutorialDokument24 SeitenChimera X TutorialMaruf Ahmed BhuiyanNoch keine Bewertungen
- Guidelines and QuestionsDokument6 SeitenGuidelines and Questionsd nNoch keine Bewertungen
- Pymol TutorialDokument21 SeitenPymol TutorialpravindshawNoch keine Bewertungen
- Swiss Deep View PDFDokument10 SeitenSwiss Deep View PDFKarthi ShanmugamNoch keine Bewertungen
- Tutorial PymolDokument16 SeitenTutorial PymolfahraniNoch keine Bewertungen
- PyRx Tutorial CheatSheet ALPerryman72010Dokument4 SeitenPyRx Tutorial CheatSheet ALPerryman72010Constantine แนืหะฟืะรืำNoch keine Bewertungen
- Pymol PDFDokument28 SeitenPymol PDFLucas MouraNoch keine Bewertungen
- PyMOL TutorialDokument7 SeitenPyMOL TutorialMatheus SilvaNoch keine Bewertungen
- PyMol PracticalDokument7 SeitenPyMol PracticalTom FlemingNoch keine Bewertungen
- Cinelerra-GG Quick Start Guide: 1 - Install The SoftwareDokument8 SeitenCinelerra-GG Quick Start Guide: 1 - Install The SoftwareJehova Villa MartinezNoch keine Bewertungen
- Discovery Studio 2.1 Tutorials GuideDokument130 SeitenDiscovery Studio 2.1 Tutorials Guidedark_geneNoch keine Bewertungen
- PyMOL QuickRef 0905Dokument4 SeitenPyMOL QuickRef 0905scsynNoch keine Bewertungen
- Assignment 1ADokument12 SeitenAssignment 1AianNoch keine Bewertungen
- HighScore Plus GuideDokument19 SeitenHighScore Plus Guidejompa21Noch keine Bewertungen
- Apsim UI IntroductionDokument39 SeitenApsim UI IntroductionHerlina UtamiNoch keine Bewertungen
- OS15 ManualDokument283 SeitenOS15 ManualUBD LIBRARYNoch keine Bewertungen
- Quick Start RocFall TutorialDokument13 SeitenQuick Start RocFall TutorialMARCONoch keine Bewertungen
- Logger Pro InstructionsDokument3 SeitenLogger Pro InstructionsIlmuncMakesuillNoch keine Bewertungen
- Short Manual To Chemdraw: The Chemical Drawing ProgramDokument22 SeitenShort Manual To Chemdraw: The Chemical Drawing ProgramiDuckyNoch keine Bewertungen
- Protein Structure ExplorationDokument22 SeitenProtein Structure Explorationsuveer698Noch keine Bewertungen
- Helicon FocusDokument24 SeitenHelicon FocuspaoloregarNoch keine Bewertungen
- Pymol Tutorial3 PDFDokument39 SeitenPymol Tutorial3 PDFThe Negative AngleNoch keine Bewertungen
- Pymol Tutorial: Part of Biochemistry 712 and Biochemistry 660Dokument39 SeitenPymol Tutorial: Part of Biochemistry 712 and Biochemistry 660Debanjan GoswamiNoch keine Bewertungen
- Intro Py MOLDokument29 SeitenIntro Py MOLsplash166Noch keine Bewertungen
- JMP IntroductionDokument5 SeitenJMP IntroductionLee WintersNoch keine Bewertungen
- Powder X-Ray Diffraction Patterns Using Bruker D8 Adv GADDS InstrumentDokument14 SeitenPowder X-Ray Diffraction Patterns Using Bruker D8 Adv GADDS InstrumentChau MaiNoch keine Bewertungen
- You Are What You Eat Project: Step by Step TutorialDokument12 SeitenYou Are What You Eat Project: Step by Step Tutorialapi-310416394Noch keine Bewertungen
- ICP-MS User Booklet DescargrDokument15 SeitenICP-MS User Booklet DescargrJeampier Roca CamposNoch keine Bewertungen
- Labintro Digital Image AnalysisDokument5 SeitenLabintro Digital Image AnalysiscabrahaoNoch keine Bewertungen
- Learning Activity Sheet Computer Science 2Dokument17 SeitenLearning Activity Sheet Computer Science 2Jaeda BaltazarNoch keine Bewertungen
- Basic Image Processing ImagejDokument9 SeitenBasic Image Processing ImagejSg89Noch keine Bewertungen
- An ABAQUS Tutorial by A Student For StudDokument28 SeitenAn ABAQUS Tutorial by A Student For StudsanthoshNoch keine Bewertungen
- Geomatica TutorialDokument20 SeitenGeomatica TutorialIlyas NannmarNoch keine Bewertungen
- Exercise A1 - Simple Ogee SetupDokument20 SeitenExercise A1 - Simple Ogee SetupCarlos Luis Oyuela Gomez100% (2)
- OIM Analysis Tutorials PDFDokument62 SeitenOIM Analysis Tutorials PDFArun Sundar100% (1)
- Manifestation ManualDokument6 SeitenManifestation Manualxrepus102650% (2)
- Introduction to PANalytical X’Pert HighScore Plus v3.0Dokument20 SeitenIntroduction to PANalytical X’Pert HighScore Plus v3.0Fred montalvo amancaNoch keine Bewertungen
- Photoshop-Image Editing: Opening A File: Photoshop WorkspaceDokument7 SeitenPhotoshop-Image Editing: Opening A File: Photoshop WorkspaceBłue MoønNoch keine Bewertungen
- Operating Instructions: Cary 100 UV-Vis Spectrophotometer Scan ModuleDokument1 SeiteOperating Instructions: Cary 100 UV-Vis Spectrophotometer Scan ModuleRamakrishnan HariNoch keine Bewertungen
- Skyline Targeted Method Editing TutorialDokument26 SeitenSkyline Targeted Method Editing TutorialCristian BorosNoch keine Bewertungen
- JCharts Help Share TradingDokument7 SeitenJCharts Help Share Tradingrevathichellam4601100% (1)
- ENVI Tutorial: Basic Hyperspectral AnalysisDokument10 SeitenENVI Tutorial: Basic Hyperspectral AnalysisPartha GhoshNoch keine Bewertungen
- How To Use The AxiovisionDokument21 SeitenHow To Use The AxiovisiondaneshnedaieNoch keine Bewertungen
- BrainSuite Workshop 2017 GUI David ShattuckDokument53 SeitenBrainSuite Workshop 2017 GUI David ShattuckKHAMSI ZINEBNoch keine Bewertungen
- And Introduction To Photoshop: Group IDokument51 SeitenAnd Introduction To Photoshop: Group ILyka BunuanNoch keine Bewertungen
- Quick Guide To Beam Analysis Using Strand7Dokument15 SeitenQuick Guide To Beam Analysis Using Strand7Tarek AbulailNoch keine Bewertungen
- Skyline Hi-Res MetabolomicsDokument18 SeitenSkyline Hi-Res MetabolomicsShahinuzzamanAdaNoch keine Bewertungen
- PacsCube Version 3 ContentsDokument25 SeitenPacsCube Version 3 ContentsMukesh RanaNoch keine Bewertungen
- DipTrace TutorialDokument134 SeitenDipTrace TutorialMetalloyNoch keine Bewertungen
- Lab 5Dokument13 SeitenLab 5GEO 2004Noch keine Bewertungen
- TopOpt 3D UserGuide: Getting Started with Design OptimizationDokument11 SeitenTopOpt 3D UserGuide: Getting Started with Design OptimizationAvon AltaNoch keine Bewertungen
- Unsupervised Classfication Using ER MapperDokument9 SeitenUnsupervised Classfication Using ER MapperavisenicNoch keine Bewertungen
- Multimedia ProgamsDokument10 SeitenMultimedia ProgamshafsaadnNoch keine Bewertungen
- Spice Som Guide EnglishDokument14 SeitenSpice Som Guide EnglishCao ThangNoch keine Bewertungen
- 3 (1) .7.3 Centricity PACS Quick Start Guide - ExtendedDokument17 Seiten3 (1) .7.3 Centricity PACS Quick Start Guide - ExtendedAlexandrIlinNoch keine Bewertungen
- PHOTOSHOP CS4 BASICS GUIDEDokument43 SeitenPHOTOSHOP CS4 BASICS GUIDETarek CheaibNoch keine Bewertungen
- SOP UV-VIS Spectrophotometer (Shimadzu UV-2600Dokument5 SeitenSOP UV-VIS Spectrophotometer (Shimadzu UV-2600valsquare valsquareNoch keine Bewertungen
- MILKDROP Preset Authoring GuideDokument34 SeitenMILKDROP Preset Authoring GuideMacelo CostaNoch keine Bewertungen
- The Ridiculously Simple Guide to Keynote For Mac: Creating Presentations On Your MacVon EverandThe Ridiculously Simple Guide to Keynote For Mac: Creating Presentations On Your MacNoch keine Bewertungen
- Owncloud 01Dokument1 SeiteOwncloud 01Sandeep KaushikNoch keine Bewertungen
- On Bacterial GrowthDokument4 SeitenOn Bacterial GrowthSandeep KaushikNoch keine Bewertungen
- Computational ApproachesDokument12 SeitenComputational ApproachesSandeep KaushikNoch keine Bewertungen
- Biofuels Annual - New Delhi - India - 7!10!2018Dokument26 SeitenBiofuels Annual - New Delhi - India - 7!10!2018Sandeep KaushikNoch keine Bewertungen
- A Trip To SpainDokument2 SeitenA Trip To SpainSandeep KaushikNoch keine Bewertungen
- BoardingCard 193586500 OPO WRODokument1 SeiteBoardingCard 193586500 OPO WROSandeep KaushikNoch keine Bewertungen
- Katju Statement On Odias and OdishaDokument8 SeitenKatju Statement On Odias and OdishaSandeep KaushikNoch keine Bewertungen
- Financial Economic Assessment Biodiesel Production Use India PDFDokument30 SeitenFinancial Economic Assessment Biodiesel Production Use India PDFSandeep KaushikNoch keine Bewertungen
- Fly HighDokument1 SeiteFly HighSandeep KaushikNoch keine Bewertungen
- Lead BioinformaticianDokument2 SeitenLead BioinformaticianSandeep KaushikNoch keine Bewertungen
- The Prevalence of Inappropriate Image Duplication in Biomedical Research PublicationsDokument8 SeitenThe Prevalence of Inappropriate Image Duplication in Biomedical Research PublicationsSandeep KaushikNoch keine Bewertungen
- Wonderful Seminars Jan-March 2019Dokument1 SeiteWonderful Seminars Jan-March 2019Sandeep KaushikNoch keine Bewertungen
- Text Mining and Its Potential Applications in Systems BiologyDokument9 SeitenText Mining and Its Potential Applications in Systems BiologySandeep KaushikNoch keine Bewertungen
- Internet of Things Data in Healthcare (Iots) (From Machines To Human Kind)Dokument1 SeiteInternet of Things Data in Healthcare (Iots) (From Machines To Human Kind)Sandeep KaushikNoch keine Bewertungen
- Essence of the Readily Seen Sun GodDokument235 SeitenEssence of the Readily Seen Sun GodRakesh YadavNoch keine Bewertungen
- MCD Results PDFDokument8 SeitenMCD Results PDFSandeep KaushikNoch keine Bewertungen
- Lead BioinformaticianDokument2 SeitenLead BioinformaticianSandeep KaushikNoch keine Bewertungen
- Some Spiritual Gyaan PDFDokument11 SeitenSome Spiritual Gyaan PDFSandeep KaushikNoch keine Bewertungen
- 10 1038/sj CLPT 6100235Dokument1 Seite10 1038/sj CLPT 6100235Sandeep KaushikNoch keine Bewertungen
- yESAO Exchange Program Award 2018Dokument5 SeitenyESAO Exchange Program Award 2018Sandeep KaushikNoch keine Bewertungen
- Notification Central University of Himachal Pradesh Teaching PostsDokument8 SeitenNotification Central University of Himachal Pradesh Teaching PostsSandeep KaushikNoch keine Bewertungen
- SplicingEfficiency FinalDokument2 SeitenSplicingEfficiency FinalSandeep KaushikNoch keine Bewertungen
- VMD: Visual Molecular Dynamics: William Humphrey, Andrew Dalke, and Klaus SchultenDokument6 SeitenVMD: Visual Molecular Dynamics: William Humphrey, Andrew Dalke, and Klaus SchultenSandeep KaushikNoch keine Bewertungen
- Should Indian Researchers Pay To Get Their Work PublishedDokument11 SeitenShould Indian Researchers Pay To Get Their Work PublishedSandeep KaushikNoch keine Bewertungen
- 264 4 FullDokument3 Seiten264 4 FullSandeep KaushikNoch keine Bewertungen
- XML TutorialDokument17 SeitenXML TutorialNic Hansen100% (2)
- VersionDokument1 SeiteVersionSandeep KaushikNoch keine Bewertungen
- Adobe Acrobat Xi Merge PDF Files Tutorial Ue PDFDokument1 SeiteAdobe Acrobat Xi Merge PDF Files Tutorial Ue PDFSandeep KaushikNoch keine Bewertungen
- S R Res Nov 16Dokument18 SeitenS R Res Nov 16Sandeep KaushikNoch keine Bewertungen
- Article in Press: Process Safety and Environmental ProtectionDokument12 SeitenArticle in Press: Process Safety and Environmental ProtectionAbeer El ShahawyNoch keine Bewertungen
- Dwnload Full Medical Terminology For Health Professions 7th Edition Ehrlich Test Bank PDFDokument36 SeitenDwnload Full Medical Terminology For Health Professions 7th Edition Ehrlich Test Bank PDFmarchingpalmerbabg100% (9)
- Biochemical TestDokument11 SeitenBiochemical Testz707yNoch keine Bewertungen
- IGCSE Biology IntroductionDokument21 SeitenIGCSE Biology IntroductionSilya ZhaoNoch keine Bewertungen
- Acceptability: ©WHO/Sergey VolkovDokument26 SeitenAcceptability: ©WHO/Sergey VolkovBeatriz PatricioNoch keine Bewertungen
- (1983) Stratum CorneumDokument275 Seiten(1983) Stratum CorneumfauzNoch keine Bewertungen
- Language Isolates and Their Genetic IdenDokument3 SeitenLanguage Isolates and Their Genetic IdenGirawa KandaNoch keine Bewertungen
- Manganese Metallurgy Part IDokument23 SeitenManganese Metallurgy Part IRichard Jesus VillagarayNoch keine Bewertungen
- Understanding Morphology Second EditionDokument14 SeitenUnderstanding Morphology Second EditionJulia TamNoch keine Bewertungen
- Daniel Dennett - Kinds of MindsDokument130 SeitenDaniel Dennett - Kinds of MindsRamona Anisie100% (1)
- Sterilization Validation GuideDokument10 SeitenSterilization Validation GuideVenkata Rama50% (2)
- A2 Biology Notes For EdexcelDokument150 SeitenA2 Biology Notes For EdexcelAbdulrahman Jijawi83% (6)
- Articles About LinguisticsDokument14 SeitenArticles About LinguisticslinhhkNoch keine Bewertungen
- Experiment 5 - General and Specific Tests For CarbohydratesDokument18 SeitenExperiment 5 - General and Specific Tests For CarbohydratesArthur Lorenz Paraguison100% (1)
- Excretory System of FrogDokument3 SeitenExcretory System of FrogSree Lekshmi V SNoch keine Bewertungen
- Adenium PropogationDokument1 SeiteAdenium PropogationTanNoch keine Bewertungen
- Non Linear PharmacokineticsDokument33 SeitenNon Linear PharmacokineticsClaudNoch keine Bewertungen
- Agricultural Science Class 3 QuestionsDokument3 SeitenAgricultural Science Class 3 QuestionsEmmanuel DominicNoch keine Bewertungen
- Emma - First Grade Literacy Case StudyDokument8 SeitenEmma - First Grade Literacy Case Studyphoebe arriane gammadNoch keine Bewertungen
- Four New Species of RasboraDokument25 SeitenFour New Species of RasboraSubhadra LaimayumNoch keine Bewertungen
- Sampling and Laboratory Analysis ProceduresDokument22 SeitenSampling and Laboratory Analysis ProceduresFauzan HardiNoch keine Bewertungen
- Biomechanics of BoneDokument27 SeitenBiomechanics of BoneJimmy ThomasNoch keine Bewertungen
- OCSPP Harmonized Test Guidelines - Master List: Available Electronically atDokument20 SeitenOCSPP Harmonized Test Guidelines - Master List: Available Electronically atmelimeli106Noch keine Bewertungen
- Cepheids.: No. of Systematic Area Position Variables DeviationDokument9 SeitenCepheids.: No. of Systematic Area Position Variables DeviationLefrina GusrianiiNoch keine Bewertungen
- Expression and ExtractionDokument34 SeitenExpression and ExtractionNaveenNoch keine Bewertungen
- Quarterly Journal of Experimental Physiology - 1908 - HerringDokument39 SeitenQuarterly Journal of Experimental Physiology - 1908 - HerringdikhsyaguragainNoch keine Bewertungen
- Skema Kertas 1&2 N9 2020Dokument25 SeitenSkema Kertas 1&2 N9 2020Azween SabtuNoch keine Bewertungen
- Lab Report Enzyme LabDokument7 SeitenLab Report Enzyme LabrualrightNoch keine Bewertungen
- SPEXSP Handbook2016Dokument126 SeitenSPEXSP Handbook2016dchyNoch keine Bewertungen