Das könnte Ihnen auch gefallen
- BioinformaticsDokument10 SeitenBioinformaticsapi-398565393100% (1)
- Reflective Essay.1Dokument31 SeitenReflective Essay.1Nora Lyn M. TorresNoch keine Bewertungen
- Database DesignDokument10 SeitenDatabase DesignAishath NajaathNoch keine Bewertungen
- Implement Windows Server DNSDokument13 SeitenImplement Windows Server DNSJorge Condor AguilarNoch keine Bewertungen
- SpringBoot All ProgramsDokument16 SeitenSpringBoot All ProgramsHitesh WadhwaniNoch keine Bewertungen
- Presentation For UCLDokument33 SeitenPresentation For UCLabrahamaudu100% (1)
- Paul's Three Missionary Journeys PDFDokument6 SeitenPaul's Three Missionary Journeys PDFLavanya Priya Sathyan100% (1)
- Predicting Clinical Outcomes From Genomics Deep Learning ModelsDokument11 SeitenPredicting Clinical Outcomes From Genomics Deep Learning ModelsVenkat KunaNoch keine Bewertungen
- Under Ground Cable Fault Detection Using IOT-1Dokument22 SeitenUnder Ground Cable Fault Detection Using IOT-1Sarath V PradheepNoch keine Bewertungen
- Drilling Into Big Cancer-Genome DataDokument5 SeitenDrilling Into Big Cancer-Genome DataLavanya Priya SathyanNoch keine Bewertungen
- Performance Analysis and Evaluation of Different Data Mining Algorithms Used For Cancer ClassificationDokument7 SeitenPerformance Analysis and Evaluation of Different Data Mining Algorithms Used For Cancer ClassificationPAUL AKAMPURIRANoch keine Bewertungen
- Gene Selection and Classification of Microarray Data Using Convolutional Neural NetworkDokument6 SeitenGene Selection and Classification of Microarray Data Using Convolutional Neural NetworkTajbia HossainNoch keine Bewertungen
- 1 s2.0 S266660302200015X MainDokument7 Seiten1 s2.0 S266660302200015X MainSINGSTONNoch keine Bewertungen
- Develop An Extended Model of CNN Algorithm in Deep Learning For Bone Tumor Detection and Its ApplicationDokument8 SeitenDevelop An Extended Model of CNN Algorithm in Deep Learning For Bone Tumor Detection and Its ApplicationInternational Journal of Innovative Science and Research TechnologyNoch keine Bewertungen
- Best Practices For Variant Calling in Clinical Sequencing: Review Open AccessDokument13 SeitenBest Practices For Variant Calling in Clinical Sequencing: Review Open AccessAhmad SolihinNoch keine Bewertungen
- Performance Evaluation of Machine Learning Algorithms in Post-Operative Life Expectancy in The Lung Cancer PatientsDokument12 SeitenPerformance Evaluation of Machine Learning Algorithms in Post-Operative Life Expectancy in The Lung Cancer PatientsNabaNoch keine Bewertungen
- Fair Shares Building and Benefiting From Healthcare AI WithDokument4 SeitenFair Shares Building and Benefiting From Healthcare AI WithDeniz KarakullukcuNoch keine Bewertungen
- Mining Big Data: Breast Cancer Prediction Using DT - SVM Hybrid ModelDokument12 SeitenMining Big Data: Breast Cancer Prediction Using DT - SVM Hybrid ModelDr-Rabia AlmamalookNoch keine Bewertungen
- Performance Analysis of Breast Cancer Classification Using Decision Tree ClassifiersDokument7 SeitenPerformance Analysis of Breast Cancer Classification Using Decision Tree ClassifiersPAUL AKAMPURIRANoch keine Bewertungen
- Modernistic Approach To Clustering AlgorithmsDokument5 SeitenModernistic Approach To Clustering AlgorithmsInternational Journal of Innovative Science and Research TechnologyNoch keine Bewertungen
- 22-GWO Papers-27-02-2024Dokument16 Seiten22-GWO Papers-27-02-2024gunjanbhartia2003Noch keine Bewertungen
- Plagiarism1 - ReportDokument8 SeitenPlagiarism1 - ReportDeepak GourNoch keine Bewertungen
- Data Mining Framework For Network Intrusion Detection Using Efficient TechniquesDokument6 SeitenData Mining Framework For Network Intrusion Detection Using Efficient TechniquesPoonam KilaniyaNoch keine Bewertungen
- Performance Evaluation of Machine Learning Algorithms in Post-Operative Life Expectancy in The Lung Cancer PatientsDokument11 SeitenPerformance Evaluation of Machine Learning Algorithms in Post-Operative Life Expectancy in The Lung Cancer PatientsDanjuma Kwetishe JoroNoch keine Bewertungen
- Editorial: Artificial Intelligence in Medical ApplicationsDokument3 SeitenEditorial: Artificial Intelligence in Medical Applicationsabelteshe_340263389Noch keine Bewertungen
- Review Article: A Review of Feature Selection and Feature Extraction Methods Applied On Microarray DataDokument14 SeitenReview Article: A Review of Feature Selection and Feature Extraction Methods Applied On Microarray Datafahad darNoch keine Bewertungen
- Cancer Gene Detection & DiagnosisDokument7 SeitenCancer Gene Detection & DiagnosisIJRASETPublicationsNoch keine Bewertungen
- BMC Bioinformatics: Bayesian Clustering and Feature Selection For Cancer Tissue SamplesDokument18 SeitenBMC Bioinformatics: Bayesian Clustering and Feature Selection For Cancer Tissue SamplesY YNoch keine Bewertungen
- A Microarray Gene Expression Data Classification UDokument14 SeitenA Microarray Gene Expression Data Classification UHuseyin OztoprakNoch keine Bewertungen
- Heart PredictionDokument6 SeitenHeart PredictionArunkumarNoch keine Bewertungen
- Analysis of Heart Disease Using in Data Mining Tools Orange and WekaDokument7 SeitenAnalysis of Heart Disease Using in Data Mining Tools Orange and WekabekNoch keine Bewertungen
- Biological Data Mining: Back GroundDokument3 SeitenBiological Data Mining: Back GroundaysiinNoch keine Bewertungen
- Barbosa 2018Dokument26 SeitenBarbosa 2018Bikash Ranjan SamalNoch keine Bewertungen
- Expert Systems With Applications: Huey Fang Ong, Norwati Mustapha, Hazlina Hamdan, Rozita Rosli, Aida MustaphaDokument18 SeitenExpert Systems With Applications: Huey Fang Ong, Norwati Mustapha, Hazlina Hamdan, Rozita Rosli, Aida MustaphaAhmed AlesaNoch keine Bewertungen
- Diagnosis and Prognosis of Breast Cancer Using Multi Classification AlgorithmDokument5 SeitenDiagnosis and Prognosis of Breast Cancer Using Multi Classification AlgorithmEditor IJRITCCNoch keine Bewertungen
- Analysis of Classification Techniques For Medical Data: April 2018Dokument6 SeitenAnalysis of Classification Techniques For Medical Data: April 2018Ahmed MostafaNoch keine Bewertungen
- Research Paper On Brain Tumor DetectionDokument8 SeitenResearch Paper On Brain Tumor Detectiongz8jpg31100% (1)
- Cutting-Edge Technologies That Could Reshape HTM and Health ITDokument6 SeitenCutting-Edge Technologies That Could Reshape HTM and Health ITsripriyakrishnaNoch keine Bewertungen
- CRISP-MED-DM A Methodology of Diagnosing Breast CancerDokument13 SeitenCRISP-MED-DM A Methodology of Diagnosing Breast CancerInternational Journal of Innovative Science and Research TechnologyNoch keine Bewertungen
- Shaking Up The Value ChainDokument7 SeitenShaking Up The Value ChainPaparazzi Agencia CreativaNoch keine Bewertungen
- Expert Systems With Applications: Bichen Zheng, Sang Won Yoon, Sarah S. LamDokument7 SeitenExpert Systems With Applications: Bichen Zheng, Sang Won Yoon, Sarah S. LamKashishNoch keine Bewertungen
- Feature Selection For Breast Cancer Detection Using Machine Learning AlgorithmsDokument4 SeitenFeature Selection For Breast Cancer Detection Using Machine Learning AlgorithmsAnjumNoch keine Bewertungen
- Cervical Cancer Cell PredictionDokument5 SeitenCervical Cancer Cell PredictionEngineering and Scientific International JournalNoch keine Bewertungen
- An Application of Machine Learning in Ivf Comparing The Accuracy of Classification Alogithims For The Prediction of TwinDokument5 SeitenAn Application of Machine Learning in Ivf Comparing The Accuracy of Classification Alogithims For The Prediction of TwinRajeeNoch keine Bewertungen
- Prediction of Breast Cancer Disease Using Decision Tree AlgorithmDokument6 SeitenPrediction of Breast Cancer Disease Using Decision Tree AlgorithmMelkamu100% (1)
- NTTTTTDokument5 SeitenNTTTTTDippal IsraniNoch keine Bewertungen
- Detection of Oral Cancer Using Deep Neural Based Adaptive Fuzzy System in Data Mining TechniquesDokument8 SeitenDetection of Oral Cancer Using Deep Neural Based Adaptive Fuzzy System in Data Mining TechniquesHarisankar BNoch keine Bewertungen
- Scientific Software: Seeing The Snps Between Us: Technology FeatureDokument6 SeitenScientific Software: Seeing The Snps Between Us: Technology Featuresalman672003Noch keine Bewertungen
- Practical Pattern MatchingDokument4 SeitenPractical Pattern MatchingDanteANoch keine Bewertungen
- Oncology Nursing Trends and Issues 5Dokument9 SeitenOncology Nursing Trends and Issues 5DaichiNoch keine Bewertungen
- Extraction and Processing of Medical Data Using Data Mining TechniquesDokument5 SeitenExtraction and Processing of Medical Data Using Data Mining TechniquesInternational Journal of Innovative Science and Research TechnologyNoch keine Bewertungen
- Astuti 2018 J. Phys. Conf. Ser. 971 012003Dokument8 SeitenAstuti 2018 J. Phys. Conf. Ser. 971 012003fauzieahmad526Noch keine Bewertungen
- 2013 Article 6555Dokument9 Seiten2013 Article 6555Erez CohenNoch keine Bewertungen
- Advanced Sequencing Technologies: Methods and Goals: Nature Reviews Genetics June 2004Dokument12 SeitenAdvanced Sequencing Technologies: Methods and Goals: Nature Reviews Genetics June 2004Leyvert De JesusNoch keine Bewertungen
- Case of Dose Regimen Optimization For Paclitaxel Case StudyDokument2 SeitenCase of Dose Regimen Optimization For Paclitaxel Case Studyshahpriyanka26Noch keine Bewertungen
- Breast Cancer Classification Using Machine LearningDokument9 SeitenBreast Cancer Classification Using Machine LearningVinothNoch keine Bewertungen
- Standardization in Analytical LabsDokument7 SeitenStandardization in Analytical LabsMatthew KleeNoch keine Bewertungen
- Visualizing Transformers For Breast HistopathologyDokument8 SeitenVisualizing Transformers For Breast HistopathologyInternational Journal of Innovative Science and Research TechnologyNoch keine Bewertungen
- Forensic Science International: Genetics: Yao-Yuan Liu, Sallyann HarbisonDokument12 SeitenForensic Science International: Genetics: Yao-Yuan Liu, Sallyann HarbisonMylenaNoch keine Bewertungen
- Biomolecules 12 01133Dokument15 SeitenBiomolecules 12 01133Srikanth ShenoyNoch keine Bewertungen
- Recent Advances and Emerging Applications in Text and Data Mining For Biomedical DiscoveryDokument10 SeitenRecent Advances and Emerging Applications in Text and Data Mining For Biomedical Discoveryanish.t.pNoch keine Bewertungen
- Students' Admission Prediction Using GRBST With Distributed Data MiningDokument5 SeitenStudents' Admission Prediction Using GRBST With Distributed Data MiningshridharbombNoch keine Bewertungen
- Thesis Brain Tumor DetectionDokument6 SeitenThesis Brain Tumor Detectionracheljohnstondesmoines100% (2)
- Investigating Reproducibility and Tracking ProvenaDokument15 SeitenInvestigating Reproducibility and Tracking ProvenaAhmad SolihinNoch keine Bewertungen
- Multi-Channel Deep Convolutional Neural Networks For Multi-Classifying Thyroid DiseaseDokument14 SeitenMulti-Channel Deep Convolutional Neural Networks For Multi-Classifying Thyroid DiseaseRihab BEN LAMINENoch keine Bewertungen
- A Comparative Study For Brain Tumor Detection Analysis Using CNN and VGG-16 and Its ApplicationDokument8 SeitenA Comparative Study For Brain Tumor Detection Analysis Using CNN and VGG-16 and Its ApplicationInternational Journal of Innovative Science and Research TechnologyNoch keine Bewertungen
- Integration and Visualization of Gene Selection and Gene Regulatory Networks for Cancer GenomeVon EverandIntegration and Visualization of Gene Selection and Gene Regulatory Networks for Cancer GenomeNoch keine Bewertungen
- 21 53 1 PBDokument15 Seiten21 53 1 PBLavanya Priya SathyanNoch keine Bewertungen
- Research and Reviews: Journal of Agriculture and Allied SciencesDokument4 SeitenResearch and Reviews: Journal of Agriculture and Allied SciencesLavanya Priya SathyanNoch keine Bewertungen
- Antidiabetic Effect of Syzygium Cumini L. Seed On Type 2 Diabetic RatsDokument8 SeitenAntidiabetic Effect of Syzygium Cumini L. Seed On Type 2 Diabetic RatsLavanya Priya Sathyan100% (1)
- Available Online Through: WWW - Jpronline.infoDokument7 SeitenAvailable Online Through: WWW - Jpronline.infoLavanya Priya SathyanNoch keine Bewertungen
- 7205 28711 1 PB PDFDokument12 Seiten7205 28711 1 PB PDFLavanya Priya SathyanNoch keine Bewertungen
- Biological Activity of Some Fruit Leaves and Chemical Investigation On Dillenia Indica LeavesDokument6 SeitenBiological Activity of Some Fruit Leaves and Chemical Investigation On Dillenia Indica LeavesLavanya Priya SathyanNoch keine Bewertungen
- 7430 9561 1 PB PDFDokument4 Seiten7430 9561 1 PB PDFLavanya Priya SathyanNoch keine Bewertungen
- Structural Investigations of Some Bi O Based GlassesDokument8 SeitenStructural Investigations of Some Bi O Based GlassesLavanya Priya SathyanNoch keine Bewertungen
- 116 428 1 PBDokument16 Seiten116 428 1 PBLavanya Priya SathyanNoch keine Bewertungen
- Format 1 PDFDokument9 SeitenFormat 1 PDFLavanya Priya SathyanNoch keine Bewertungen
- Chapter Proposal Submission FormDokument3 SeitenChapter Proposal Submission FormLavanya Priya SathyanNoch keine Bewertungen
- My Diabetes Reversal Diet Plan: 50 ML Coconut Milk + 100 ML Water + Piece of Ginger + 1 Sachet Stevia. About 65 Kcal EaDokument1 SeiteMy Diabetes Reversal Diet Plan: 50 ML Coconut Milk + 100 ML Water + Piece of Ginger + 1 Sachet Stevia. About 65 Kcal EaLavanya Priya SathyanNoch keine Bewertungen
- Interesting Facts About TimothyDokument3 SeitenInteresting Facts About TimothyLavanya Priya Sathyan100% (1)
- DA3 Unit 1 Assessment: NameDokument4 SeitenDA3 Unit 1 Assessment: NameLavanya Priya SathyanNoch keine Bewertungen
- Kumon of Matthews Tuition Increase Letter 2018Dokument1 SeiteKumon of Matthews Tuition Increase Letter 2018Lavanya Priya SathyanNoch keine Bewertungen
- Prayer Points For RevivalDokument1 SeitePrayer Points For RevivalLavanya Priya SathyanNoch keine Bewertungen
- Introduction To Password Cracking Part 1Dokument8 SeitenIntroduction To Password Cracking Part 1Tibyan MuhammedNoch keine Bewertungen
- Input and Output DevicesDokument2 SeitenInput and Output DevicesRonalyn Mae LagmayNoch keine Bewertungen
- Slides By: Andrew Stephenson Georgia Gwinnett CollegeDokument22 SeitenSlides By: Andrew Stephenson Georgia Gwinnett CollegeRamya GunukulaNoch keine Bewertungen
- Abe 14 Activity Module 4Dokument9 SeitenAbe 14 Activity Module 4Erman DeanoNoch keine Bewertungen
- Free Autocad For Students 3-18-2020 RevisedDokument4 SeitenFree Autocad For Students 3-18-2020 RevisedJorge GuerreroNoch keine Bewertungen
- Sandys SlidesCarnivalDokument40 SeitenSandys SlidesCarnivalEman KhanNoch keine Bewertungen
- CSE 3D Printing ReportDokument18 SeitenCSE 3D Printing ReportAksh RawalNoch keine Bewertungen
- STR-A6100 Series Data Sheet: Off-Line PRC Controllers With Integrated Power MOSFETDokument25 SeitenSTR-A6100 Series Data Sheet: Off-Line PRC Controllers With Integrated Power MOSFETRaja RajaNoch keine Bewertungen
- Cisco MDS 9000 Family NX-OS Fabric Configuration GuideDokument278 SeitenCisco MDS 9000 Family NX-OS Fabric Configuration Guidenanganam5491Noch keine Bewertungen
- DBM CSC FormDokument4 SeitenDBM CSC FormJing Goal Merit0% (1)
- Powerline Laundry CatalogDokument32 SeitenPowerline Laundry CatalogRudy prastamaNoch keine Bewertungen
- Product Brochure HP X58045Dokument17 SeitenProduct Brochure HP X58045terNoch keine Bewertungen
- Rup Vs ScrumDokument9 SeitenRup Vs ScrumMehdi AmiriNoch keine Bewertungen
- Massive Open On-Line and Formats of MediaDokument8 SeitenMassive Open On-Line and Formats of MediaPatricia CondeNoch keine Bewertungen
- Last Clean ExceptionDokument4 SeitenLast Clean ExceptionHavanna G.Noch keine Bewertungen
- CST363 Final Exam HuyNguyenDokument9 SeitenCST363 Final Exam HuyNguyenHuy NguyenNoch keine Bewertungen
- Faza 2Dokument6 SeitenFaza 2MIHAIL ALEXANDRU MOLDOVANNoch keine Bewertungen
- Deep Learning For Human Beings v2Dokument110 SeitenDeep Learning For Human Beings v2Anya NieveNoch keine Bewertungen
- 400, 800, 2300, 2300D Series: Small, Two-Piece, Nonmetallic Raceway SystemsDokument8 Seiten400, 800, 2300, 2300D Series: Small, Two-Piece, Nonmetallic Raceway SystemsMohanathan VCNoch keine Bewertungen
- Swift Programming LanguageDokument14 SeitenSwift Programming LanguagepharezeNoch keine Bewertungen
- DP-A120 User GuideDokument92 SeitenDP-A120 User GuideCarlos Raga100% (5)
- Tcontwebbac01 IomDokument70 SeitenTcontwebbac01 IomWagner antonio rabelo costaNoch keine Bewertungen
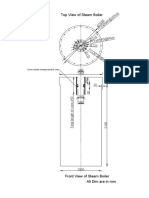
- Top View of Steam Boiler: PCDØ270@6NosDokument1 SeiteTop View of Steam Boiler: PCDØ270@6NosAnkur SinghNoch keine Bewertungen
- Structural Products & Systems: Toolkit 8 Design Software User Guide For The Republic of IrelandDokument3 SeitenStructural Products & Systems: Toolkit 8 Design Software User Guide For The Republic of IrelandDass DassNoch keine Bewertungen