Das könnte Ihnen auch gefallen
- Delta Cad ManualDokument0 SeitenDelta Cad ManualAndrei PantuNoch keine Bewertungen
- Presentation On NTPC Inspection ServicesDokument34 SeitenPresentation On NTPC Inspection ServicesAshwani Dogra100% (3)
- The Technical Writing ProcessDokument37 SeitenThe Technical Writing ProcessEzekiel D. RodriguezNoch keine Bewertungen
- AOL Student Guide PDFDokument84 SeitenAOL Student Guide PDFbanavaram1Noch keine Bewertungen
- Project Schedule Management GuideDokument33 SeitenProject Schedule Management GuideABDELMALEK .TEBOUDANoch keine Bewertungen
- Usage of Permits in SAP Plant Maintenance - SAP Blogs PDFDokument25 SeitenUsage of Permits in SAP Plant Maintenance - SAP Blogs PDFvishal naikNoch keine Bewertungen
- Orca Manual 5 0 0Dokument1.317 SeitenOrca Manual 5 0 0Ferzz Montejo50% (2)
- Business FunctionsDokument88 SeitenBusiness Functionsmurali dhanunjay0% (2)
- Kwant 1.3.1 Documentation: C. W. Groth, M. Wimmer, A. R. Akhmerov, X. Waintal, Et AlDokument169 SeitenKwant 1.3.1 Documentation: C. W. Groth, M. Wimmer, A. R. Akhmerov, X. Waintal, Et AlFerzz MontejoNoch keine Bewertungen
- 2012 05 Neese ComputationalChemistryDokument129 Seiten2012 05 Neese ComputationalChemistryBis ChemNoch keine Bewertungen
- KotlerDokument9 SeitenKotlerGitanjali BaggaNoch keine Bewertungen
- Backpropagation: Fundamentals and Applications for Preparing Data for Training in Deep LearningVon EverandBackpropagation: Fundamentals and Applications for Preparing Data for Training in Deep LearningNoch keine Bewertungen
- Jump-Start Guide for ORCA Quantum Chemistry SuiteDokument23 SeitenJump-Start Guide for ORCA Quantum Chemistry SuiteJoel Montalvo Acosta100% (1)
- CTF Series Binary ExploitationDokument51 SeitenCTF Series Binary ExploitationPhilemon SundayNoch keine Bewertungen
- Worksharing and Parallel LoopsDokument23 SeitenWorksharing and Parallel LoopsMOHAMMED HANZLA HUSSAINNoch keine Bewertungen
- Lecture Open MPDokument25 SeitenLecture Open MPDr. V. Padmavathi Associate ProfessorNoch keine Bewertungen
- IPSL Boot Camp Part 5: CDO and NCO: 1 Climate Data OperatorsDokument10 SeitenIPSL Boot Camp Part 5: CDO and NCO: 1 Climate Data OperatorsMd CassimNoch keine Bewertungen
- 07 OpenMPDokument28 Seiten07 OpenMPHamid KishaNoch keine Bewertungen
- Openmp: Dr. Nitya Hariharan (Intel)Dokument42 SeitenOpenmp: Dr. Nitya Hariharan (Intel)KiaraNoch keine Bewertungen
- W8L1 OpenMP5 SynchronizationDokument20 SeitenW8L1 OpenMP5 Synchronizationl215376Noch keine Bewertungen
- Quick TourDokument7 SeitenQuick Tourdidodad248Noch keine Bewertungen
- Unit 4 Shared-Memory Parallel Programming With OpenmpDokument37 SeitenUnit 4 Shared-Memory Parallel Programming With OpenmpSudha PalaniNoch keine Bewertungen
- OpenMP for Parallel ProcessingDokument40 SeitenOpenMP for Parallel ProcessingEngr. Sohaib JamalNoch keine Bewertungen
- OMP Common Core-VossDokument217 SeitenOMP Common Core-VossAvinashNoch keine Bewertungen
- W8L2 OpenMP6 FurthertopicsDokument20 SeitenW8L2 OpenMP6 Furthertopicsl215376Noch keine Bewertungen
- 4 OpenmpDokument32 Seiten4 Openmpgirishcherry12Noch keine Bewertungen
- Case Study: CFD Dr. Graham Pullan University of Cambridge: Nvidia TeslaDokument56 SeitenCase Study: CFD Dr. Graham Pullan University of Cambridge: Nvidia Teslafengwang2102Noch keine Bewertungen
- Introduction To OpenMPDokument22 SeitenIntroduction To OpenMPVariable 14Noch keine Bewertungen
- Programming the qbcan and Sensing the WorldDokument10 SeitenProgramming the qbcan and Sensing the WorldiguanajazzNoch keine Bewertungen
- Module 2 - New 1Dokument72 SeitenModule 2 - New 1Bantu AadhfNoch keine Bewertungen
- Avrbeginners 04 Jumps Calls and The Stack 1.0.1Dokument11 SeitenAvrbeginners 04 Jumps Calls and The Stack 1.0.1PintuabcNoch keine Bewertungen
- Chap4 OpenMPDokument35 SeitenChap4 OpenMPMichael ShiNoch keine Bewertungen
- Manual Stack To STamPSDokument8 SeitenManual Stack To STamPSgwisnuNoch keine Bewertungen
- Gaussian & GaussView Software SuiteDokument70 SeitenGaussian & GaussView Software Suite顧浩Noch keine Bewertungen
- Atom-4 2 0Dokument17 SeitenAtom-4 2 0Elias BritoNoch keine Bewertungen
- Open MP 2016Dokument45 SeitenOpen MP 2016MitoneNoch keine Bewertungen
- Ejercicio 1 3Dokument17 SeitenEjercicio 1 3SamuelNoch keine Bewertungen
- UNEX GIS WSAnalysis AdvArc10 Python PDFDokument34 SeitenUNEX GIS WSAnalysis AdvArc10 Python PDFgokonkNoch keine Bewertungen
- Fundamentals of Machine Learning Support Vector Machines, Practical SessionDokument4 SeitenFundamentals of Machine Learning Support Vector Machines, Practical SessionvothiquynhyenNoch keine Bewertungen
- File 1741Dokument10 SeitenFile 1741Renan DanielNoch keine Bewertungen
- Curs09 Dtur Working With Gaussian09Dokument64 SeitenCurs09 Dtur Working With Gaussian09Ewerton CaetanoNoch keine Bewertungen
- OpenMP ExamplesDokument12 SeitenOpenMP ExamplesFernando Montoya CubasNoch keine Bewertungen
- Verilog Coding Guideline: Author: TrumenDokument51 SeitenVerilog Coding Guideline: Author: TrumenNK NKNoch keine Bewertungen
- Xe 62011 Open MPDokument46 SeitenXe 62011 Open MPJulianMoralesNoch keine Bewertungen
- 1B02 - Bring Your Python To The NSO Circus - NSO DevDays 2017Dokument12 Seiten1B02 - Bring Your Python To The NSO Circus - NSO DevDays 2017Ala JebnounNoch keine Bewertungen
- Module - II ADokument94 SeitenModule - II APamusainagaharshavardhanNoch keine Bewertungen
- Abinitio-MaterialDokument11 SeitenAbinitio-Materialinderjeetkumar singhNoch keine Bewertungen
- Concurrent Programiing Tutorial-2Dokument8 SeitenConcurrent Programiing Tutorial-2niku007Noch keine Bewertungen
- Open MP2Dokument28 SeitenOpen MP2l215376Noch keine Bewertungen
- Num TechDokument39 SeitenNum Techandres pythonNoch keine Bewertungen
- PostProcessing BruyereDokument45 SeitenPostProcessing BruyereThirugnanam DhandayuthapaniNoch keine Bewertungen
- ML Lab 11 Manual - Neural Networks (Ver4)Dokument8 SeitenML Lab 11 Manual - Neural Networks (Ver4)dodela6303Noch keine Bewertungen
- Lab Manual: 1 Dept of E&C, PACE, MangaloreDokument78 SeitenLab Manual: 1 Dept of E&C, PACE, MangaloreMr.Mohammed Zakir B ELECTRONICS & COMMUNICATIONNoch keine Bewertungen
- Utf 8''week4Dokument15 SeitenUtf 8''week4devendra416Noch keine Bewertungen
- Code Generation Issues: Compilation 2014Dokument19 SeitenCode Generation Issues: Compilation 2014finiNoch keine Bewertungen
- CPMD 2Dokument32 SeitenCPMD 2Anonymous 0pBRsQXfnxNoch keine Bewertungen
- Threads APIDokument16 SeitenThreads APIMinh Quân ĐoànNoch keine Bewertungen
- Lecture17 12Dokument86 SeitenLecture17 12Amal HajarNoch keine Bewertungen
- Programming Shared-Memory Platforms With Openmp: John Mellor-CrummeyDokument46 SeitenProgramming Shared-Memory Platforms With Openmp: John Mellor-CrummeyaskbilladdmicrosoftNoch keine Bewertungen
- S3 L4 Pa1 PDFDokument94 SeitenS3 L4 Pa1 PDFShani ArainNoch keine Bewertungen
- CASACalibrationSISS 2014 PDFDokument181 SeitenCASACalibrationSISS 2014 PDFarijit mannaNoch keine Bewertungen
- Yambo Quick GuideDokument7 SeitenYambo Quick Guidehantarto5844Noch keine Bewertungen
- Quantum Espresso Module 2 Walkthrough: H2 Molecule Energy and Geometry OptimizationDokument10 SeitenQuantum Espresso Module 2 Walkthrough: H2 Molecule Energy and Geometry OptimizationIsmael Antonio Gonzalez RamirezNoch keine Bewertungen
- OpenGL ARB Vertex Program OverviewDokument58 SeitenOpenGL ARB Vertex Program OverviewBalqis YafisNoch keine Bewertungen
- CS553 Homework #3: Benchmarking StorageDokument5 SeitenCS553 Homework #3: Benchmarking StorageHariharan ShankarNoch keine Bewertungen
- Vlsi LabDokument161 SeitenVlsi LabNaveen Kumar RavillaNoch keine Bewertungen
- Open MPDokument35 SeitenOpen MPDebarshi MajumderNoch keine Bewertungen
- CAMB LectureDokument46 SeitenCAMB LectureJhon J Galan RNoch keine Bewertungen
- DLMCR Project ReportDokument14 SeitenDLMCR Project ReportGovindNoch keine Bewertungen
- Georglm: Software For Generalised Linear Spatial Models Using RDokument46 SeitenGeorglm: Software For Generalised Linear Spatial Models Using RGloria AcostaNoch keine Bewertungen
- Radial Basis Networks: Fundamentals and Applications for The Activation Functions of Artificial Neural NetworksVon EverandRadial Basis Networks: Fundamentals and Applications for The Activation Functions of Artificial Neural NetworksNoch keine Bewertungen
- CSC Spring School 2018: Orca 4.0 & GabeditDokument59 SeitenCSC Spring School 2018: Orca 4.0 & GabeditFerzz MontejoNoch keine Bewertungen
- I GormanDokument2.358 SeitenI GormanFerzz MontejoNoch keine Bewertungen
- User Guide PDFDokument27 SeitenUser Guide PDFCarlitos TapiaNoch keine Bewertungen
- E 0343028041Dokument14 SeitenE 0343028041International Journal of computational Engineering research (IJCER)Noch keine Bewertungen
- Ug PDFDokument265 SeitenUg PDFHyakansahsNoch keine Bewertungen
- Vesta ManualDokument177 SeitenVesta ManualLuis JimenezNoch keine Bewertungen
- MultiwfnDokument395 SeitenMultiwfnFerzz MontejoNoch keine Bewertungen
- Lecture OrcaDokument74 SeitenLecture Orcayeuchem040404Noch keine Bewertungen
- Gabedit 2.10 - ManualDokument24 SeitenGabedit 2.10 - ManualHenrique JuniorNoch keine Bewertungen
- Gabedit 2.10 - ManualDokument24 SeitenGabedit 2.10 - ManualHenrique JuniorNoch keine Bewertungen
- Siesta-4 0Dokument144 SeitenSiesta-4 0Renato BertoniNoch keine Bewertungen
- Siesta-4 0Dokument144 SeitenSiesta-4 0Renato BertoniNoch keine Bewertungen
- Lecture OrcaDokument74 SeitenLecture Orcayeuchem040404Noch keine Bewertungen
- DCACPsDokument57 SeitenDCACPsFerzz MontejoNoch keine Bewertungen
- User Defined ProgrammingDokument33 SeitenUser Defined ProgrammingSaravanan PalanisamyNoch keine Bewertungen
- Nciplot ManualDokument16 SeitenNciplot ManualFerzz MontejoNoch keine Bewertungen
- OrcaGabedit ManualDokument35 SeitenOrcaGabedit ManualFerzz MontejoNoch keine Bewertungen
- Manual WFNDokument385 SeitenManual WFNFerzz MontejoNoch keine Bewertungen
- Chemical Doping of Graphene c0jm02922jDokument12 SeitenChemical Doping of Graphene c0jm02922jFerzz MontejoNoch keine Bewertungen
- Siesta Trunk ManualDokument141 SeitenSiesta Trunk ManualFerzz MontejoNoch keine Bewertungen
- Cubic FormulaDokument9 SeitenCubic FormulaIlija CrnacNoch keine Bewertungen
- D. Null OperatorDokument49 SeitenD. Null OperatorPlayIt All100% (2)
- AmeriCom - NEC SV8100 WebPro Doc - V1Dokument48 SeitenAmeriCom - NEC SV8100 WebPro Doc - V1Dante Fajardo OlmosNoch keine Bewertungen
- Difference between supercomputers and mainframe computersDokument5 SeitenDifference between supercomputers and mainframe computersHafeez NiaziNoch keine Bewertungen
- Function QuizDokument1 SeiteFunction QuizAnonymous VKSwhG1c100% (1)
- Lane 3000Dokument2 SeitenLane 3000guerramag1Noch keine Bewertungen
- Xtrade Website DemoDokument72 SeitenXtrade Website Demoapi-3704498Noch keine Bewertungen
- Cs 6456-Object Oriented Programming-Faq UNIT-I (2 Marks)Dokument12 SeitenCs 6456-Object Oriented Programming-Faq UNIT-I (2 Marks)Balasubramani ManickamNoch keine Bewertungen
- Ajax For MobilityDokument2 SeitenAjax For MobilitymachinelearnerNoch keine Bewertungen
- DSP Group ProjectDokument2 SeitenDSP Group ProjectMuhammad LuqmanNoch keine Bewertungen
- Itu-G 168Dokument134 SeitenItu-G 168Ilya FinkelNoch keine Bewertungen
- hAND OFFDokument22 SeitenhAND OFFjenath1100% (1)
- Philippine ICT Coordinator DesignationDokument2 SeitenPhilippine ICT Coordinator DesignationJOHNBERGIN E. MACARAEGNoch keine Bewertungen
- BVN Update FormDokument1 SeiteBVN Update FormTharj Imam50% (2)
- Java MCQS ExplainedDokument20 SeitenJava MCQS ExplainedM Husnain ShahidNoch keine Bewertungen
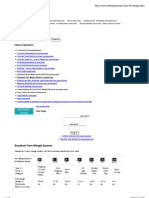
- Standard Yarn Weight SystemDokument2 SeitenStandard Yarn Weight SystemsignemrNoch keine Bewertungen
- Ac2000 WebDokument192 SeitenAc2000 WebmahirouxNoch keine Bewertungen
- Ajeet Singh - ResumeDokument4 SeitenAjeet Singh - ResumeTaslimNoch keine Bewertungen
- SAP Tables OtherDokument15 SeitenSAP Tables OthervtolkyNoch keine Bewertungen
- Oracle Istore Order ManagementDokument4 SeitenOracle Istore Order ManagementDikshaNoch keine Bewertungen
- Professional Surveillance Software ManualDokument63 SeitenProfessional Surveillance Software ManualJaime Andrés Sarria CastilloNoch keine Bewertungen
- DullGelinasWheeler9e TB Chapter 03Dokument18 SeitenDullGelinasWheeler9e TB Chapter 03JimboWineNoch keine Bewertungen