Das könnte Ihnen auch gefallen
- Bariwal 2013Dokument21 SeitenBariwal 2013Le Anh NhatNoch keine Bewertungen
- Handbook of Coordination Catalysis in Organic ChemistryVon EverandHandbook of Coordination Catalysis in Organic ChemistryNoch keine Bewertungen
- Rethinking Amide Bond SynthesisDokument9 SeitenRethinking Amide Bond SynthesisPaolo SuatingNoch keine Bewertungen
- Chemfiles Vol. 9, No. 1 - MIDA-protected Boronate EstersDokument12 SeitenChemfiles Vol. 9, No. 1 - MIDA-protected Boronate EstersSigma-AldrichNoch keine Bewertungen
- The Negishi Coupling: An Update - Aldrichimica Acta Vol. 38 No. 3Dokument48 SeitenThe Negishi Coupling: An Update - Aldrichimica Acta Vol. 38 No. 3Sigma Aldrich Chemistry100% (1)
- Strategies for Palladium-Catalyzed Non-directed and Directed C bond H Bond FunctionalizationVon EverandStrategies for Palladium-Catalyzed Non-directed and Directed C bond H Bond FunctionalizationAnant R. KapdiNoch keine Bewertungen
- Oxidation of Organic Compounds: Medium Effects in Radical ReactionsVon EverandOxidation of Organic Compounds: Medium Effects in Radical ReactionsBewertung: 4 von 5 Sternen4/5 (1)
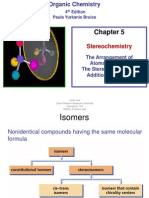
- Stereochemistry: 4 Edition Paula Yurkanis BruiceDokument43 SeitenStereochemistry: 4 Edition Paula Yurkanis Bruicenrguerrerod100% (1)
- Functionalization of Indole and Pyrrole Cores Via Michael - Type AdditionsDokument61 SeitenFunctionalization of Indole and Pyrrole Cores Via Michael - Type AdditionsSilvina CancianNoch keine Bewertungen
- Organic Chemistry: Stereospecificity and StereoselectivityDokument10 SeitenOrganic Chemistry: Stereospecificity and StereoselectivityNida Adrees100% (1)
- A) Explain Why Thioesters Are Used As Chain Extenders Instead of Oxygen Esters in The Acetate PathwayDokument11 SeitenA) Explain Why Thioesters Are Used As Chain Extenders Instead of Oxygen Esters in The Acetate Pathwayzacks100% (1)
- Reference Book For CSIR-UGC-NET-GATE ChemistryDokument2 SeitenReference Book For CSIR-UGC-NET-GATE Chemistryprasant906100% (1)
- 210 Fa 15 Exam 2 KEYDokument19 Seiten210 Fa 15 Exam 2 KEYdsarathy1Noch keine Bewertungen
- C C, C N, C O CouplingDokument67 SeitenC C, C N, C O CouplingAnonymous vRpzQ2BLNoch keine Bewertungen
- Carbenes NitrenesDokument29 SeitenCarbenes NitrenesManish KaushalNoch keine Bewertungen
- Heterocyclic Chemistry: Parts 2 and 3: Year 3, Semester 1 DR Boa, C120b, A.n.boa@hull - Ac.ukDokument22 SeitenHeterocyclic Chemistry: Parts 2 and 3: Year 3, Semester 1 DR Boa, C120b, A.n.boa@hull - Ac.ukKike MenesesNoch keine Bewertungen
- Organometallic CompoundsDokument66 SeitenOrganometallic CompoundsJon Ho100% (1)
- Org Chem Sem 3 Paper 2Dokument15 SeitenOrg Chem Sem 3 Paper 2Rohit DeshmukhNoch keine Bewertungen
- Green Chemistry CHP 1-17Dokument59 SeitenGreen Chemistry CHP 1-17Basu GargNoch keine Bewertungen
- Retrosynthetic Analysis PDFDokument6 SeitenRetrosynthetic Analysis PDFNoleNoch keine Bewertungen
- Green Synthesis of Silver Nanoparticles Using Azadirachta Indica Aqueous Leaf ExtractDokument8 SeitenGreen Synthesis of Silver Nanoparticles Using Azadirachta Indica Aqueous Leaf Extractdear mishayyamNoch keine Bewertungen
- Coordination Chemistry IV: Reactions & MechanismsDokument23 SeitenCoordination Chemistry IV: Reactions & MechanismsMagspie SamNoch keine Bewertungen
- B.sc. (Chemistry)Dokument41 SeitenB.sc. (Chemistry)RahulNoch keine Bewertungen
- Inorg Reactionn MechanismsDokument38 SeitenInorg Reactionn MechanismsThabang MolekoNoch keine Bewertungen
- Homogeneous Catalysis With Metal Phosphine ComplexesDokument494 SeitenHomogeneous Catalysis With Metal Phosphine ComplexesLiliana CapulínNoch keine Bewertungen
- SCH 206-Carboxylic Acids PDFDokument48 SeitenSCH 206-Carboxylic Acids PDFShivani DamorNoch keine Bewertungen
- Organometallic Compunds: D. JIM LIVINGSTON, Asst - Prof in Chemistry, ST - John's College, PalaiDokument23 SeitenOrganometallic Compunds: D. JIM LIVINGSTON, Asst - Prof in Chemistry, ST - John's College, PalaiJim LivingstonNoch keine Bewertungen
- Synthesis of AzidesDokument72 SeitenSynthesis of AzidesTanner WarehamNoch keine Bewertungen
- Neighbouring Group Participation or NGP inDokument4 SeitenNeighbouring Group Participation or NGP inbharatbhushansankhya100% (1)
- Retrosynthesis Approach For Natural ProductsDokument9 SeitenRetrosynthesis Approach For Natural ProductsSantosh ButleNoch keine Bewertungen
- Isolobal AnalogyDokument4 SeitenIsolobal Analogyindu priyaNoch keine Bewertungen
- Chalcone To Pyrimidine by Urea Indian PaperDokument7 SeitenChalcone To Pyrimidine by Urea Indian PaperAnkit Kumar Singh100% (1)
- Grignard Reagents Review MeetingDokument28 SeitenGrignard Reagents Review MeetingShivali SharmaNoch keine Bewertungen
- Mono - and Bis-N-heterocyclic Carbene ComplexesDokument6 SeitenMono - and Bis-N-heterocyclic Carbene Complexesnepertorres6169Noch keine Bewertungen
- Synthesis of Lactones and LactamsDokument1.093 SeitenSynthesis of Lactones and LactamsnestorNoch keine Bewertungen
- Reductions by The Alumino - and Borohydrides in Organic SynthesisDokument236 SeitenReductions by The Alumino - and Borohydrides in Organic Synthesisjfjd6889100% (1)
- Compendium On Problems in Physical-Organic ChemistryDokument27 SeitenCompendium On Problems in Physical-Organic ChemistryHaryokoe buzzNoch keine Bewertungen
- Synthesis of Benzimidazole Using Boric AcidDokument5 SeitenSynthesis of Benzimidazole Using Boric Acidg20kpNoch keine Bewertungen
- Multicomponent Reactions - Ambhaikar (July 2004)Dokument6 SeitenMulticomponent Reactions - Ambhaikar (July 2004)muopioidreceptorNoch keine Bewertungen
- Practice Questions-Conformational AnalysisDokument4 SeitenPractice Questions-Conformational AnalysisHarry Zgambo100% (1)
- Chap6 Free Radical PolymnDokument64 SeitenChap6 Free Radical PolymnsanjeevpmNoch keine Bewertungen
- Demethylation With LiCl-DMF (JMolCatA-Chemical2007)Dokument8 SeitenDemethylation With LiCl-DMF (JMolCatA-Chemical2007)Archawin_mooNoch keine Bewertungen
- Comparing The SN1 and SN2 Reactions - Master Organic ChemistryDokument5 SeitenComparing The SN1 and SN2 Reactions - Master Organic Chemistryprince ranaNoch keine Bewertungen
- Organic Chemistry: Richard F. Daley and Sally J. DaleyDokument48 SeitenOrganic Chemistry: Richard F. Daley and Sally J. DaleyJuan Carlos CamaleNoch keine Bewertungen
- Contents of All Volumes: Natural Products Structural Diversity-I Secondary Metabolites: Organization and BiosynthesisDokument755 SeitenContents of All Volumes: Natural Products Structural Diversity-I Secondary Metabolites: Organization and BiosynthesisMarcio BragaNoch keine Bewertungen
- In Situ Click ChemistryDokument2 SeitenIn Situ Click ChemistryClara CarreraNoch keine Bewertungen
- Structure and Properties of Inorganic Solids: International Series of Monographs in Solid State PhysicsVon EverandStructure and Properties of Inorganic Solids: International Series of Monographs in Solid State PhysicsNoch keine Bewertungen
- MSCCH07Dokument385 SeitenMSCCH07capdesuro100% (1)
- Lab ManualDokument19 SeitenLab Manualanon_467104036Noch keine Bewertungen
- Coordination Chemistry—XVI: XVIth International Conference on Coordination ChemistryVon EverandCoordination Chemistry—XVI: XVIth International Conference on Coordination ChemistryNoch keine Bewertungen
- Introduction To Medicinal Green ChemistryDokument12 SeitenIntroduction To Medicinal Green ChemistryAnonymous rm2rf6Noch keine Bewertungen
- Rajender S. Varma - Solvent-Free Organic Syntheses Using Supported Reagents and Microwave IrradiationDokument13 SeitenRajender S. Varma - Solvent-Free Organic Syntheses Using Supported Reagents and Microwave IrradiationnnnnjwNoch keine Bewertungen
- Green Chemistry by Nanocatalysis PDFDokument12 SeitenGreen Chemistry by Nanocatalysis PDFAnonymous rm2rf6Noch keine Bewertungen
- PNAS Article PDFDokument6 SeitenPNAS Article PDFAnonymous rm2rf6Noch keine Bewertungen
- Emily Moore: Undergraduate AssistantDokument2 SeitenEmily Moore: Undergraduate Assistantapi-546234388Noch keine Bewertungen
- Summary of 2018 Dental Exam Requirements: Required SectionsDokument13 SeitenSummary of 2018 Dental Exam Requirements: Required Sectionsjentothesky100% (1)
- EssayDokument6 SeitenEssayapi-285104197Noch keine Bewertungen
- Components To The Teacher: What Is Top Notch? ActiveteachDokument1 SeiteComponents To The Teacher: What Is Top Notch? ActiveteachMikelyn AndersonNoch keine Bewertungen
- Pedigree Analysis WorksheetDokument6 SeitenPedigree Analysis WorksheetLloaana 12Noch keine Bewertungen
- Microsoft Solution Acceleration Hack PlaybookDokument11 SeitenMicrosoft Solution Acceleration Hack PlaybookDaniel SilvaNoch keine Bewertungen
- Activity Learning Sheets in Oral - Com - TYPES OF SPEECH CONTEXT - Guinsaugon High SchoolDokument5 SeitenActivity Learning Sheets in Oral - Com - TYPES OF SPEECH CONTEXT - Guinsaugon High SchoolCatzMacedaMeekNoch keine Bewertungen
- Phil-IRI POST TEST Reading Selection Form A4Dokument6 SeitenPhil-IRI POST TEST Reading Selection Form A4ariel mateo monesNoch keine Bewertungen
- Lesson Plan in Physical EducationDokument4 SeitenLesson Plan in Physical EducationGemmalyn DeVilla De CastroNoch keine Bewertungen
- New Text DocumentDokument3 SeitenNew Text DocumentmenuselectNoch keine Bewertungen
- Public Policy PDFDokument525 SeitenPublic Policy PDFMutemwa Emmanuel Tafira93% (15)
- Startup Media List New 2020Dokument33 SeitenStartup Media List New 2020Ashita RawatNoch keine Bewertungen
- Pe GradesDokument6 SeitenPe Gradesarvin bermudezNoch keine Bewertungen
- Hnology and Livelihood Education: Quarter 1 - Module 3: CookeryDokument28 SeitenHnology and Livelihood Education: Quarter 1 - Module 3: CookeryDomeng Karo100% (2)
- Is Lacans Theory of The Mirror Stage Still ValidDokument10 SeitenIs Lacans Theory of The Mirror Stage Still ValidEmin MammadovNoch keine Bewertungen
- Wa0004.Dokument4 SeitenWa0004.Paras GuptaNoch keine Bewertungen
- A Review On Computational Methods Based On Machine Learning and Deep Learning Techniques For Malaria DetectionDokument5 SeitenA Review On Computational Methods Based On Machine Learning and Deep Learning Techniques For Malaria DetectionGopi ChandNoch keine Bewertungen
- Literature Review Template: Writing CentreDokument1 SeiteLiterature Review Template: Writing CentreSyed Imam BakharNoch keine Bewertungen
- HRD-QOHSER-002 Application FormDokument7 SeitenHRD-QOHSER-002 Application FormMajestik ggNoch keine Bewertungen
- LP - Fire HazardsDokument4 SeitenLP - Fire Hazardsrexson de villaNoch keine Bewertungen
- Observation Sheet TemplateDokument4 SeitenObservation Sheet TemplateSebastian GorhamNoch keine Bewertungen
- 49 Commencement Exercises Senior High School Department S.Y. 2018 - 2019Dokument8 Seiten49 Commencement Exercises Senior High School Department S.Y. 2018 - 2019rey anne danoNoch keine Bewertungen
- The One Who Is Not Loyal To The Motherland... Can Never Be Loyal To Anyone... - KushDokument5 SeitenThe One Who Is Not Loyal To The Motherland... Can Never Be Loyal To Anyone... - KushanonymousNoch keine Bewertungen
- Asha NRHM HryDokument8 SeitenAsha NRHM Hrynareshjangra397Noch keine Bewertungen
- The Future of The Humanities and Liberal Arts in A Global AgeDokument30 SeitenThe Future of The Humanities and Liberal Arts in A Global AgeJohn McAteer100% (1)
- AQA GCSE Maths Foundation Paper 1 Mark Scheme November 2022Dokument30 SeitenAQA GCSE Maths Foundation Paper 1 Mark Scheme November 2022CCSC124-Soham MaityNoch keine Bewertungen
- Types of ReadingDokument5 SeitenTypes of ReadingfatinfiqahNoch keine Bewertungen
- Assignment/ Tugasan - Health and Wellness 2Dokument14 SeitenAssignment/ Tugasan - Health and Wellness 2Metran Electrical & ITNoch keine Bewertungen
- 1-Article Text-1-1-10-20200210Dokument156 Seiten1-Article Text-1-1-10-20200210manafNoch keine Bewertungen
- English IV: 3 Partial Week 4Dokument17 SeitenEnglish IV: 3 Partial Week 4Luis Manuel GonzálezNoch keine Bewertungen