Das könnte Ihnen auch gefallen
- Applied Electrostatics (ICAES 2004): Proceedings of the Fifth International Conference on Applied ElectrostaticsVon EverandApplied Electrostatics (ICAES 2004): Proceedings of the Fifth International Conference on Applied ElectrostaticsNoch keine Bewertungen
- Multi-Functional Thermosensitive Composite Microspheres With High Magnetic Susceptibility Based On Magnetite Colloidal Nanoparticle ClustersDokument9 SeitenMulti-Functional Thermosensitive Composite Microspheres With High Magnetic Susceptibility Based On Magnetite Colloidal Nanoparticle ClustersKashifa KhushiNoch keine Bewertungen
- A Versatile Method For Grafting Polymers On Nanoparticles: John P. Gann and Mingdi YanDokument5 SeitenA Versatile Method For Grafting Polymers On Nanoparticles: John P. Gann and Mingdi YanJames WallaceNoch keine Bewertungen
- Synthesis, Thermal Stability and Properties of Zno NanoparticlesDokument6 SeitenSynthesis, Thermal Stability and Properties of Zno NanoparticlessadhuNoch keine Bewertungen
- Particle Size Determination Using TEMDokument11 SeitenParticle Size Determination Using TEMAlan TiradoNoch keine Bewertungen
- Gold Nanoparticle Effects in Polymerase Chain ReacDokument7 SeitenGold Nanoparticle Effects in Polymerase Chain ReacMyat OoNoch keine Bewertungen
- Size Control of Gold Nanocrystals in Citrate ReductionDokument10 SeitenSize Control of Gold Nanocrystals in Citrate ReductionJosé Adriano SilvaNoch keine Bewertungen
- 19 AgDokument4 Seiten19 AgDeepikaNoch keine Bewertungen
- Magnetic Beads For ECEDokument5 SeitenMagnetic Beads For ECEQian WanNoch keine Bewertungen
- Wang 2009Dokument6 SeitenWang 2009tapashrautrayNoch keine Bewertungen
- Applied Surface Science: A.W. Tan, R. Ismail, K.H. Chua, R. Ahmad, S.A. Akbar, B. Pingguan-MurphyDokument10 SeitenApplied Surface Science: A.W. Tan, R. Ismail, K.H. Chua, R. Ahmad, S.A. Akbar, B. Pingguan-Murphyey_mieNoch keine Bewertungen
- Stodart 34Dokument15 SeitenStodart 34Ana Maria Moreno PenaNoch keine Bewertungen
- Preparation and Physisorption Characterization of - Glucose-Templated Mesoporous Silica Sol-Gel MaterialsDokument7 SeitenPreparation and Physisorption Characterization of - Glucose-Templated Mesoporous Silica Sol-Gel MaterialsDwiNoch keine Bewertungen
- Veronika Kozlovskaya Et Al - Ultrathin Layer-by-Layer Hydrogels With Incorporated Gold Nanorods As pH-Sensitive Optical MaterialsDokument13 SeitenVeronika Kozlovskaya Et Al - Ultrathin Layer-by-Layer Hydrogels With Incorporated Gold Nanorods As pH-Sensitive Optical MaterialsYlpkasoNoch keine Bewertungen
- Cell Adhesion Morphology and Biochemistry On Nanotopographic Oxidized Silicon SurfacesDokument16 SeitenCell Adhesion Morphology and Biochemistry On Nanotopographic Oxidized Silicon Surfaces健康新體驗Noch keine Bewertungen
- Zinc Glycolate: A Precursor To Zno: Jaykrushna Das, Ivana R. Evans, and Deepa KhushalaniDokument3 SeitenZinc Glycolate: A Precursor To Zno: Jaykrushna Das, Ivana R. Evans, and Deepa KhushalanijeffersonNoch keine Bewertungen
- Inorganic Cluster As Single SourceDokument10 SeitenInorganic Cluster As Single Sourceprakush01975225403Noch keine Bewertungen
- Green Phosphorescence of Zinc Sulfide Optical CeraDokument12 SeitenGreen Phosphorescence of Zinc Sulfide Optical CerahulyaNoch keine Bewertungen
- Mechanistic Investigation Into Antibacterial Behaviour of Suspensions of ZnO Nanoparticles Against E CliDokument13 SeitenMechanistic Investigation Into Antibacterial Behaviour of Suspensions of ZnO Nanoparticles Against E CliFENFOGNoch keine Bewertungen
- 005 Keiji MorokumaDokument43 Seiten005 Keiji MorokumaMuraleetharan BoopathiNoch keine Bewertungen
- MacKenzie 71 1Dokument5 SeitenMacKenzie 71 1Bruno Alex Cisterna FuentesNoch keine Bewertungen
- Ijms 23 05001 v2Dokument21 SeitenIjms 23 05001 v2Felix PrawiraNoch keine Bewertungen
- Ag Decahedral ParticleDokument5 SeitenAg Decahedral ParticleHEET MAHENDRA GALANoch keine Bewertungen
- Estimation of Lattice Strain in ZnO NanoparticlesDokument12 SeitenEstimation of Lattice Strain in ZnO NanoparticleszuleNoch keine Bewertungen
- Time-Resolved SAXS Study of Nucleation and Growth of Silica ColloidsDokument4 SeitenTime-Resolved SAXS Study of Nucleation and Growth of Silica ColloidsJoeNoch keine Bewertungen
- Tufenkji2004 - Slow and Fast DepoDokument11 SeitenTufenkji2004 - Slow and Fast DepoAKHILESH PASWANNoch keine Bewertungen
- Nanoparticle Filtration by Electrospun Polymer FibersDokument9 SeitenNanoparticle Filtration by Electrospun Polymer FibersAndrésAlfaroCerazoNoch keine Bewertungen
- Soares 2014Dokument6 SeitenSoares 2014kionnysNoch keine Bewertungen
- Carbon Nanotube Arrays BasedDokument10 SeitenCarbon Nanotube Arrays Basedtunv2003Noch keine Bewertungen
- An Examination of The Growth Kinetics of - Arginine Trifluoroacetate (LATF) Crystals From Induction Period and Atomic Force Microscopy InvestigationsDokument6 SeitenAn Examination of The Growth Kinetics of - Arginine Trifluoroacetate (LATF) Crystals From Induction Period and Atomic Force Microscopy InvestigationsJayaram KrishnanNoch keine Bewertungen
- Maria Vernanda LimaDokument12 SeitenMaria Vernanda LimaSusi SusantiNoch keine Bewertungen
- Structure-Performance Correlations in Vapor Phase Deposited Self-Assembled Nanodielectrics For Organic Field-Effect TransistorsDokument11 SeitenStructure-Performance Correlations in Vapor Phase Deposited Self-Assembled Nanodielectrics For Organic Field-Effect TransistorsKomodoDSNoch keine Bewertungen
- Nano GoldDokument10 SeitenNano Gold'Amalia' Choirin SyavawiNoch keine Bewertungen
- R. Jerald Vijay Et Al. Journal of Crystal Growth 312 (2010) 420-425Dokument25 SeitenR. Jerald Vijay Et Al. Journal of Crystal Growth 312 (2010) 420-425jerryremacNoch keine Bewertungen
- Applied Surface Science: Sahely Saha, Ravi Kumar, Krishna Pramanik, Amit BiswasDokument14 SeitenApplied Surface Science: Sahely Saha, Ravi Kumar, Krishna Pramanik, Amit Biswassaheli sahaNoch keine Bewertungen
- Platonic Gold Nanocrystals : Franklin Kim, Stephen Connor, Hyunjoon Song, Tevye Kuykendall, and Peidong YangDokument5 SeitenPlatonic Gold Nanocrystals : Franklin Kim, Stephen Connor, Hyunjoon Song, Tevye Kuykendall, and Peidong YangheydaripostNoch keine Bewertungen
- Coatings 10 00491Dokument17 SeitenCoatings 10 00491JulioNoch keine Bewertungen
- Bonding MCEDokument6 SeitenBonding MCETrung NguyenNoch keine Bewertungen
- Solvothermal Synthesis and Photocatalytic Properties of ZnO Micro/nanostructures2019Dokument19 SeitenSolvothermal Synthesis and Photocatalytic Properties of ZnO Micro/nanostructures2019karina CruzNoch keine Bewertungen
- For Comlt NewayDokument5 SeitenFor Comlt NewayDIckNoch keine Bewertungen
- Editorial Hydrothermal Synthesis of NanomaterialsDokument3 SeitenEditorial Hydrothermal Synthesis of NanomaterialsMagenta MediatamaNoch keine Bewertungen
- Demir Sky I 2011Dokument8 SeitenDemir Sky I 2011Victor TeixeiraNoch keine Bewertungen
- Zno NanorodsDokument5 SeitenZno NanorodsmirelamanteamirelaNoch keine Bewertungen
- Synopsis of The Thesis Submitted To MANONMANIAM SUNDARANAR UNIVERSITY For The Award of The Degree ofDokument4 SeitenSynopsis of The Thesis Submitted To MANONMANIAM SUNDARANAR UNIVERSITY For The Award of The Degree oframNoch keine Bewertungen
- Ref 1Dokument5 SeitenRef 1Diego Alejandro Roa CalaNoch keine Bewertungen
- Santosh Kumar AIP Con Proc (2011)Dokument3 SeitenSantosh Kumar AIP Con Proc (2011)Tarun YadavNoch keine Bewertungen
- Journal of Physics: Conference Series: August 2011Dokument8 SeitenJournal of Physics: Conference Series: August 2011Satria Yudha WirawanNoch keine Bewertungen
- Nlo Crystal Growth ThesisDokument7 SeitenNlo Crystal Growth Thesislorieharrishuntsville100% (1)
- Experimental Determination of The ExtincDokument7 SeitenExperimental Determination of The Extinctoniewayne0Noch keine Bewertungen
- Siegfried 2004Dokument4 SeitenSiegfried 2004Colín Poblete BaezaNoch keine Bewertungen
- Distorted F.C.C. Arrangement of Gold Nanoclusters: A Model of Spherical Particles With Microstrains and Stacking FaultsDokument11 SeitenDistorted F.C.C. Arrangement of Gold Nanoclusters: A Model of Spherical Particles With Microstrains and Stacking FaultsAlessandro LongoNoch keine Bewertungen
- Materials Science and Engineering ADokument6 SeitenMaterials Science and Engineering ALuis Armando Hernandez MolinaNoch keine Bewertungen
- 2005 - Kho Et Al. - Deposition Method For Preparing SERS-Active Gold Nanoparticle Substrates - Analytical ChemistryDokument10 Seiten2005 - Kho Et Al. - Deposition Method For Preparing SERS-Active Gold Nanoparticle Substrates - Analytical ChemistryClaudio BiaginiNoch keine Bewertungen
- Impact of P3HT Materials Properties and Layer Architecture On OPV DeviceDokument17 SeitenImpact of P3HT Materials Properties and Layer Architecture On OPV DeviceFab AstroNoch keine Bewertungen
- Alexey Y. Koyfman, Sergei N. Magonov and Norbert O. Reich - Self-Assembly of DNA Arrays Into Multilayer StacksDokument7 SeitenAlexey Y. Koyfman, Sergei N. Magonov and Norbert O. Reich - Self-Assembly of DNA Arrays Into Multilayer StacksYopghm698Noch keine Bewertungen
- Venu JCG2 2010Dokument6 SeitenVenu JCG2 2010soma_venuNoch keine Bewertungen
- Separation of Single-Walled Carbon Nanotubes by Use of Ionic Liquid-Aided Capillary ElectrophoresisDokument8 SeitenSeparation of Single-Walled Carbon Nanotubes by Use of Ionic Liquid-Aided Capillary ElectrophoresisAlessandro PollinaNoch keine Bewertungen
- Viscosity Measurements On Colloidal Dispersions (Nanofluids) For Heat Transfer ApplicationsDokument7 SeitenViscosity Measurements On Colloidal Dispersions (Nanofluids) For Heat Transfer ApplicationsDaniel MontalvoNoch keine Bewertungen
- Nanoparticles ThesisDokument8 SeitenNanoparticles Thesisvsiqooxff100% (2)
- 3-Enhanced Magnetization in CoFe2O4 Through Hydrogen DopingDokument8 Seiten3-Enhanced Magnetization in CoFe2O4 Through Hydrogen Dopingr ZhNoch keine Bewertungen
- Chemical Engineering ScienceDokument7 SeitenChemical Engineering ScienceVishakha GaurNoch keine Bewertungen
- More TechniquesDokument15 SeitenMore TechniquesVishakha GaurNoch keine Bewertungen
- More TechniquesDokument15 SeitenMore TechniquesVishakha GaurNoch keine Bewertungen
- Thin-Film Reactors: Symbols (See AlsoDokument9 SeitenThin-Film Reactors: Symbols (See AlsoVishakha GaurNoch keine Bewertungen
- Upwind Schemes For The Wave Equation in Secon 2012 Journal of ComputationalDokument36 SeitenUpwind Schemes For The Wave Equation in Secon 2012 Journal of ComputationalVishakha GaurNoch keine Bewertungen
- Van Dorp - JohanDokument42 SeitenVan Dorp - JohanVishakha GaurNoch keine Bewertungen
- Two Scale Numerical Solution of The Electromagnetic T 2007 Mathematics and CDokument5 SeitenTwo Scale Numerical Solution of The Electromagnetic T 2007 Mathematics and CVishakha GaurNoch keine Bewertungen
- In-Situ: Characterization of Heterogeneous Catalysts Themed IssueDokument4 SeitenIn-Situ: Characterization of Heterogeneous Catalysts Themed IssueVishakha GaurNoch keine Bewertungen
- In Situ and Ex Situ Characterization: Studies of Transition Metal Containing Nanoporous CatalystsDokument216 SeitenIn Situ and Ex Situ Characterization: Studies of Transition Metal Containing Nanoporous CatalystsVishakha GaurNoch keine Bewertungen
- 10.1023/a 1019100925300Dokument7 Seiten10.1023/a 1019100925300Vishakha GaurNoch keine Bewertungen
- Erosion Calculation Using FluentDokument15 SeitenErosion Calculation Using FluentharpreetNoch keine Bewertungen
- Acscatal 5b00858 PDFDokument5 SeitenAcscatal 5b00858 PDFVishakha GaurNoch keine Bewertungen
- In Situ Raman Spectroscopy Studies of CatalystsDokument7 SeitenIn Situ Raman Spectroscopy Studies of CatalystsVishakha GaurNoch keine Bewertungen
- 11Dokument23 Seiten11Vishakha GaurNoch keine Bewertungen
- Chaubey 2019Dokument38 SeitenChaubey 2019ranim najibNoch keine Bewertungen
- 69 RNC Owners e F 0Dokument24 Seiten69 RNC Owners e F 0Frank GrenierNoch keine Bewertungen
- AGARD-AR-323 Experimental Analytical Methods Pipe Ramjet PDFDokument106 SeitenAGARD-AR-323 Experimental Analytical Methods Pipe Ramjet PDFmaurizio.desio4992Noch keine Bewertungen
- DQE SimplifiedDokument8 SeitenDQE SimplifiedBen100% (1)
- Outflow PerformanceDokument2 SeitenOutflow PerformanceMuhammad MujahidNoch keine Bewertungen
- Definitions and MCQs of Ninth Class Chemistry (Solution and Suspension)Dokument6 SeitenDefinitions and MCQs of Ninth Class Chemistry (Solution and Suspension)Sajid AliNoch keine Bewertungen
- Flow Through NozzlesDokument13 SeitenFlow Through NozzlesBharat SharmaNoch keine Bewertungen
- Sap ConcDokument192 SeitenSap ConcJose SanchezNoch keine Bewertungen
- Buettner, Becker - PCI Manual For The Design of Hollow Core Slabs (2000)Dokument141 SeitenBuettner, Becker - PCI Manual For The Design of Hollow Core Slabs (2000)DavorPijavicaNoch keine Bewertungen
- Chemical Equilibrium-2Dokument13 SeitenChemical Equilibrium-2MUHAMMAD YASEENNoch keine Bewertungen
- Magnetic Particle Test Inspection Free NDT Sample ProcedureDokument5 SeitenMagnetic Particle Test Inspection Free NDT Sample ProcedurefluitekNoch keine Bewertungen
- Mathalino: Problem 655 - Beam Deflection by Conjugate Beam MethodDokument3 SeitenMathalino: Problem 655 - Beam Deflection by Conjugate Beam MethodMd.matiur RahmanNoch keine Bewertungen
- Smart Textiles - Technology and Application Fields: Dipl.-Ing. Klaus ScheulenDokument24 SeitenSmart Textiles - Technology and Application Fields: Dipl.-Ing. Klaus Scheulentextile028Noch keine Bewertungen
- Importance of Dimensionless Numbers in Mass TransferDokument27 SeitenImportance of Dimensionless Numbers in Mass TransferAli AmmarNoch keine Bewertungen
- Grade 7 Science FinalDokument19 SeitenGrade 7 Science FinalHazel Escobio Justol CahucomNoch keine Bewertungen
- Determination of Hydrocarbon Dew Point Using A Gas ChromatographDokument11 SeitenDetermination of Hydrocarbon Dew Point Using A Gas Chromatographmajid maheriNoch keine Bewertungen
- HYDRODYNAMICSDokument28 SeitenHYDRODYNAMICSSysy monmonNoch keine Bewertungen
- Exercises Reservoir Engineering II TinaDokument11 SeitenExercises Reservoir Engineering II TinaMohamed AbdallahiNoch keine Bewertungen
- Engineering Materials (UES012) School of Physics and Materials Science Tutorial Sheet No 1-2Dokument1 SeiteEngineering Materials (UES012) School of Physics and Materials Science Tutorial Sheet No 1-2Arpit Sachdeva100% (1)
- Hot Oil Systems: Chris FinchDokument14 SeitenHot Oil Systems: Chris FinchHuzefa ArsiwalaNoch keine Bewertungen
- Non Destructive Testing of Pipelines State and Methods For Its RepairingDokument19 SeitenNon Destructive Testing of Pipelines State and Methods For Its RepairingCarlos MusellaNoch keine Bewertungen
- Engineering ThermodynamicsDokument60 SeitenEngineering ThermodynamicsJeyaram KumarNoch keine Bewertungen
- Student Exploration: Limiting ReactantsDokument3 SeitenStudent Exploration: Limiting ReactantsJohn BrauswetterNoch keine Bewertungen
- F3 Phy 1Dokument12 SeitenF3 Phy 1BUKENYA BEEE-2026Noch keine Bewertungen
- Formation of Ice Water BodiesDokument1 SeiteFormation of Ice Water BodiesedeNoch keine Bewertungen
- Basics NWP and ApplicationDokument20 SeitenBasics NWP and ApplicationCasetome IdNoch keine Bewertungen
- Seminar ReportDokument19 SeitenSeminar ReportAjit8675100% (7)
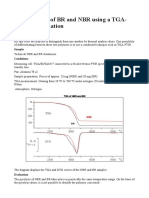
- Identification of BR and NBR Using A TGA-FTIR Combination: PurposeDokument4 SeitenIdentification of BR and NBR Using A TGA-FTIR Combination: Purposelely dwiNoch keine Bewertungen