Das könnte Ihnen auch gefallen
- Grit: The Power of Passion and PerseveranceVon EverandGrit: The Power of Passion and PerseveranceBewertung: 4 von 5 Sternen4/5 (588)
- Yi Liu, Jianfeng Xu, Mansoureh Karimiahmadabadi, Chuanzheng Zhou, and Jyoti ChattopadhyayaDokument17 SeitenYi Liu, Jianfeng Xu, Mansoureh Karimiahmadabadi, Chuanzheng Zhou, and Jyoti ChattopadhyayaDiogo DiasNoch keine Bewertungen
- The Yellow House: A Memoir (2019 National Book Award Winner)Von EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Bewertung: 4 von 5 Sternen4/5 (98)
- Vartriangle PDFDokument1 SeiteVartriangle PDFDiogo DiasNoch keine Bewertungen
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeVon EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeBewertung: 4 von 5 Sternen4/5 (5795)
- Joc - 7099Dokument8 SeitenJoc - 7099Diogo DiasNoch keine Bewertungen
- Never Split the Difference: Negotiating As If Your Life Depended On ItVon EverandNever Split the Difference: Negotiating As If Your Life Depended On ItBewertung: 4.5 von 5 Sternen4.5/5 (838)
- Joc - 7073Dokument12 SeitenJoc - 7073Diogo DiasNoch keine Bewertungen
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceVon EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceBewertung: 4 von 5 Sternen4/5 (895)
- Green Synthesis of r,β-and β,β-Dipeptides under Solvent-Free ConditionsDokument5 SeitenGreen Synthesis of r,β-and β,β-Dipeptides under Solvent-Free ConditionsDiogo DiasNoch keine Bewertungen
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersVon EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersBewertung: 4.5 von 5 Sternen4.5/5 (345)
- Theoretical Study On Conformation and Electronic State of H Uckel-Aromatic Multiply N-Confused (26) HexaphyrinsDokument11 SeitenTheoretical Study On Conformation and Electronic State of H Uckel-Aromatic Multiply N-Confused (26) HexaphyrinsDiogo DiasNoch keine Bewertungen
- Shoe Dog: A Memoir by the Creator of NikeVon EverandShoe Dog: A Memoir by the Creator of NikeBewertung: 4.5 von 5 Sternen4.5/5 (537)
- 8112 PDFDokument5 Seiten8112 PDFDiogo DiasNoch keine Bewertungen
- Joc - 7085Dokument7 SeitenJoc - 7085Diogo DiasNoch keine Bewertungen
- The Little Book of Hygge: Danish Secrets to Happy LivingVon EverandThe Little Book of Hygge: Danish Secrets to Happy LivingBewertung: 3.5 von 5 Sternen3.5/5 (400)
- Facile Synthesis of 3-Nitro-2-Substituted ThiophenesDokument5 SeitenFacile Synthesis of 3-Nitro-2-Substituted ThiophenesDiogo DiasNoch keine Bewertungen
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureVon EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureBewertung: 4.5 von 5 Sternen4.5/5 (474)
- Rhodium-Catalyzed Reductive Allylation of Conjugated Aldehydes With Allyl AcetateDokument4 SeitenRhodium-Catalyzed Reductive Allylation of Conjugated Aldehydes With Allyl AcetateDiogo DiasNoch keine Bewertungen
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryVon EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryBewertung: 3.5 von 5 Sternen3.5/5 (231)
- Jean-Philippe Pitteloud, Zun-Ting Zhang, Yong Liang, Laura Cabrera, and Stanislaw F. WnukDokument14 SeitenJean-Philippe Pitteloud, Zun-Ting Zhang, Yong Liang, Laura Cabrera, and Stanislaw F. WnukDiogo DiasNoch keine Bewertungen
- On Fire: The (Burning) Case for a Green New DealVon EverandOn Fire: The (Burning) Case for a Green New DealBewertung: 4 von 5 Sternen4/5 (74)
- Aplidiopsamine A, An Antiplasmodial Alkaloid From The Temperate Australian Ascidian, Aplidiopsis ConfluataDokument4 SeitenAplidiopsamine A, An Antiplasmodial Alkaloid From The Temperate Australian Ascidian, Aplidiopsis ConfluataDiogo DiasNoch keine Bewertungen
- The Emperor of All Maladies: A Biography of CancerVon EverandThe Emperor of All Maladies: A Biography of CancerBewertung: 4.5 von 5 Sternen4.5/5 (271)
- 8180 PDFDokument10 Seiten8180 PDFDiogo DiasNoch keine Bewertungen
- 8126 PDFDokument7 Seiten8126 PDFDiogo DiasNoch keine Bewertungen
- In Situ Trapping of Boc-2-Pyrrolidinylmethylzinc Iodide With Aryl Iodides: Direct Synthesis of 2-BenzylpyrrolidinesDokument4 SeitenIn Situ Trapping of Boc-2-Pyrrolidinylmethylzinc Iodide With Aryl Iodides: Direct Synthesis of 2-BenzylpyrrolidinesDiogo DiasNoch keine Bewertungen
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaVon EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaBewertung: 4.5 von 5 Sternen4.5/5 (266)
- 8299Dokument4 Seiten8299Diogo DiasNoch keine Bewertungen
- The Unwinding: An Inner History of the New AmericaVon EverandThe Unwinding: An Inner History of the New AmericaBewertung: 4 von 5 Sternen4/5 (45)
- Bingel - Hirsch Reactions On Non-IPR GD N@C (2 N 82 and 84)Dokument4 SeitenBingel - Hirsch Reactions On Non-IPR GD N@C (2 N 82 and 84)Diogo DiasNoch keine Bewertungen
- Team of Rivals: The Political Genius of Abraham LincolnVon EverandTeam of Rivals: The Political Genius of Abraham LincolnBewertung: 4.5 von 5 Sternen4.5/5 (234)
- Synthesis, Self-Assembly, and Charge Transporting Property of Contorted TetrabenzocoronenesDokument9 SeitenSynthesis, Self-Assembly, and Charge Transporting Property of Contorted TetrabenzocoronenesDiogo DiasNoch keine Bewertungen
- Nicholas Reactions in The Construction of Cyclohepta (De) Naphthalenes and Cyclohepta (De) Naphthalenones. The Total Synthesis of MicrostegiolDokument13 SeitenNicholas Reactions in The Construction of Cyclohepta (De) Naphthalenes and Cyclohepta (De) Naphthalenones. The Total Synthesis of MicrostegiolDiogo DiasNoch keine Bewertungen
- 8241 PDFDokument11 Seiten8241 PDFDiogo DiasNoch keine Bewertungen
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyVon EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyBewertung: 3.5 von 5 Sternen3.5/5 (2259)
- Synthesis of 4-Aryl-And 4-Alkyl-2-Silyl-1,3-Butadienes and Their Diels - Alder/Cross-Coupling ReactionsDokument11 SeitenSynthesis of 4-Aryl-And 4-Alkyl-2-Silyl-1,3-Butadienes and Their Diels - Alder/Cross-Coupling ReactionsDiogo DiasNoch keine Bewertungen
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreVon EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreBewertung: 4 von 5 Sternen4/5 (1091)
- Synthesis, Spectra, and Theoretical Investigations of The Triarylamines Based On 6 H-Indolo (2,3-b) QuinoxalineDokument12 SeitenSynthesis, Spectra, and Theoretical Investigations of The Triarylamines Based On 6 H-Indolo (2,3-b) QuinoxalineDiogo DiasNoch keine Bewertungen
- Kinetics and Mechanism of N-Boc Cleavage: Evidence of A Second-Order Dependence Upon Acid ConcentrationDokument9 SeitenKinetics and Mechanism of N-Boc Cleavage: Evidence of A Second-Order Dependence Upon Acid ConcentrationDiogo DiasNoch keine Bewertungen
- Syntheses of The Enantiomers of 1-Deoxynojirimycin and 1-Deoxyaltronojirimycin Via Chemo-And Diastereoselective Olefinic Oxidation of Unsaturated AminesDokument14 SeitenSyntheses of The Enantiomers of 1-Deoxynojirimycin and 1-Deoxyaltronojirimycin Via Chemo-And Diastereoselective Olefinic Oxidation of Unsaturated AminesDiogo DiasNoch keine Bewertungen
- Stereochemistry As A Tool in Deciphering The Processes of A Tandem Iminium Cyclization and Smiles RearrangementDokument8 SeitenStereochemistry As A Tool in Deciphering The Processes of A Tandem Iminium Cyclization and Smiles RearrangementDiogo DiasNoch keine Bewertungen
- Synthetic Approaches To Bicyclic Diazenium Salts: J. Wyman, M. I. Javed, N. Al-Bataineh, and M. BrewerDokument10 SeitenSynthetic Approaches To Bicyclic Diazenium Salts: J. Wyman, M. I. Javed, N. Al-Bataineh, and M. BrewerDiogo DiasNoch keine Bewertungen
- Lanthanum Tricyanide-Catalyzed Acyl Silane - Ketone Benzoin Additions and Kinetic Resolution of Resultant R-SilyloxyketonesDokument9 SeitenLanthanum Tricyanide-Catalyzed Acyl Silane - Ketone Benzoin Additions and Kinetic Resolution of Resultant R-SilyloxyketonesDiogo DiasNoch keine Bewertungen
- Mechanism of The Swern Oxidation: Significant Deviations From Transition State TheoryDokument12 SeitenMechanism of The Swern Oxidation: Significant Deviations From Transition State TheoryDiogo DiasNoch keine Bewertungen
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Von EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Bewertung: 4.5 von 5 Sternen4.5/5 (121)
- Oasismontaj TutorialnewDokument297 SeitenOasismontaj TutorialnewArmando Cesar Landa FerrelNoch keine Bewertungen
- BBFH107Dokument102 SeitenBBFH107lord100% (1)
- 3G Cluster Pre-OptimizationDokument41 Seiten3G Cluster Pre-OptimizationAditya NamaNoch keine Bewertungen
- Determination of Trace Sulfur Method With Non-Dispersive Gas PDFDokument4 SeitenDetermination of Trace Sulfur Method With Non-Dispersive Gas PDFRuben Perez AyoNoch keine Bewertungen
- Sense HatDokument2 SeitenSense HatRachel TaoNoch keine Bewertungen
- Electrostatics - 1: Charges and ChargingDokument90 SeitenElectrostatics - 1: Charges and ChargingCat123Noch keine Bewertungen
- Arria 10 FPGA Development Kit User Guide: Subscribe Send FeedbackDokument135 SeitenArria 10 FPGA Development Kit User Guide: Subscribe Send FeedbacklolNoch keine Bewertungen
- IIT AIEEE BITS Free Online Help and QuestionsDokument7 SeitenIIT AIEEE BITS Free Online Help and Questionsapi-3846151Noch keine Bewertungen
- A Computer-Based Optimization Method For: Archives FEB 24 1970Dokument325 SeitenA Computer-Based Optimization Method For: Archives FEB 24 1970Age MaradiagaNoch keine Bewertungen
- Geotechnical GEO5 Fem enDokument2 SeitenGeotechnical GEO5 Fem encesmanroe231Noch keine Bewertungen
- Calculating IPv4 Subnets - ANSWER - KEYDokument8 SeitenCalculating IPv4 Subnets - ANSWER - KEYPaul John QuirosNoch keine Bewertungen
- Her Body and Other Parties: StoriesVon EverandHer Body and Other Parties: StoriesBewertung: 4 von 5 Sternen4/5 (821)
- Scope and Sequence of Math CurriculumDokument2 SeitenScope and Sequence of Math CurriculumFaisal MunirNoch keine Bewertungen
- Biology Final Exam - Print - QuizizzDokument5 SeitenBiology Final Exam - Print - QuizizzrubelliteNoch keine Bewertungen
- Tutorial Sheet 2213Dokument2 SeitenTutorial Sheet 2213Mithilesh GuruNoch keine Bewertungen
- 7805Dokument2 Seiten7805pandiNoch keine Bewertungen
- Accord 1998 Auto Tranny DiagramDokument12 SeitenAccord 1998 Auto Tranny Diagramzeolite1100% (13)
- FT8 Hinson Tips For HF DXersDokument80 SeitenFT8 Hinson Tips For HF DXersIp CamNoch keine Bewertungen
- Poisson Brackets and Constants of The MotionDokument4 SeitenPoisson Brackets and Constants of The MotionPopoNoch keine Bewertungen
- Cs6302 Dbms Part ADokument16 SeitenCs6302 Dbms Part Avenkatesan.vrs1Noch keine Bewertungen
- Nünning - Literary Studies. Theories, Models and Methods (Excerpts)Dokument2 SeitenNünning - Literary Studies. Theories, Models and Methods (Excerpts)E KNoch keine Bewertungen
- KOCOM Data SheetDokument3 SeitenKOCOM Data SheetcgdemoyaNoch keine Bewertungen
- 2.power DiodesDokument15 Seiten2.power DiodesMuhammad Irfan100% (2)
- Humanities Ii (Operations Research)Dokument11 SeitenHumanities Ii (Operations Research)Swapan DeyNoch keine Bewertungen
- Inorganic Qualitative AnalysisDokument9 SeitenInorganic Qualitative AnalysisShireen SuhailNoch keine Bewertungen
- YAW - S Cross ReliefDokument5 SeitenYAW - S Cross ReliefRonaldNoch keine Bewertungen
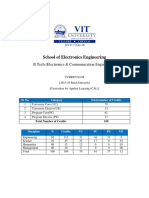
- BTechElectronicsandCommunicationEngineering CurriculumDokument7 SeitenBTechElectronicsandCommunicationEngineering CurriculumVignesh AiyerNoch keine Bewertungen
- Petrel 2013 Property Modeling Course: Module 2: Data PreparationDokument15 SeitenPetrel 2013 Property Modeling Course: Module 2: Data PreparationJaninne Campo100% (1)
- Voltaje en El EjeDokument48 SeitenVoltaje en El EjeRigoberto UrrutiaNoch keine Bewertungen
- TEMA Shell Bundle Entrance and Exit AreasDokument3 SeitenTEMA Shell Bundle Entrance and Exit AreasArunkumar MyakalaNoch keine Bewertungen
- Sully: The Untold Story Behind the Miracle on the HudsonVon EverandSully: The Untold Story Behind the Miracle on the HudsonBewertung: 4 von 5 Sternen4/5 (103)
- The Future of Geography: How the Competition in Space Will Change Our WorldVon EverandThe Future of Geography: How the Competition in Space Will Change Our WorldBewertung: 4 von 5 Sternen4/5 (6)
- The Fabric of Civilization: How Textiles Made the WorldVon EverandThe Fabric of Civilization: How Textiles Made the WorldBewertung: 4.5 von 5 Sternen4.5/5 (58)
- Hero Found: The Greatest POW Escape of the Vietnam WarVon EverandHero Found: The Greatest POW Escape of the Vietnam WarBewertung: 4 von 5 Sternen4/5 (19)
- Fire on the Horizon: The Untold Story of the Gulf Oil DisasterVon EverandFire on the Horizon: The Untold Story of the Gulf Oil DisasterNoch keine Bewertungen
- System Error: Where Big Tech Went Wrong and How We Can RebootVon EverandSystem Error: Where Big Tech Went Wrong and How We Can RebootNoch keine Bewertungen
- The Technology Trap: Capital, Labor, and Power in the Age of AutomationVon EverandThe Technology Trap: Capital, Labor, and Power in the Age of AutomationBewertung: 4.5 von 5 Sternen4.5/5 (46)