Beruflich Dokumente
Kultur Dokumente
Symposium On Neurodegeneration
Hochgeladen von
schizoid.paranoidOriginalbeschreibung:
Originaltitel
Copyright
Verfügbare Formate
Dieses Dokument teilen
Dokument teilen oder einbetten
Stufen Sie dieses Dokument als nützlich ein?
Sind diese Inhalte unangemessen?
Dieses Dokument meldenCopyright:
Verfügbare Formate
Symposium On Neurodegeneration
Hochgeladen von
schizoid.paranoidCopyright:
Verfügbare Formate
C O M M E N TA RY
Initiation and propagation of
neurodegeneration
Christian Haass1,2
Although substantial progress has is the cause of neurodegeneration? For prion cognition12. Collectively, do these studies dis-
been made in understanding the disorders, it is clear that the scrapie form of the credit the amyloid cascade hypothesis? Not
molecular and pathological bases of prion protein (PrPsc) causes neuronal death8, necessarily.
neurodegeneration, there have been but in other diseases, such as Alzheimer’s Genetic studies have shown that, in auto-
few successes in the clinic and a disease, the toxic protein or species is not as somal dominant familial Alzheimer’s dis-
number of fundamental questions evident. Moreover, the prion disease field has ease, mutations in the genes that encode the
remain unanswered. Is this skepticism an advantage in that its animal models have amyloid-β precursor protein and the two
misplaced, or do the words of Sir Isaac a defined and clear endpoint: death. Recent presenilins (PS1 and PS2) alter Aβ genera-
Newton hold true, that “what we know is advances with organotypic brain slices even tion and strongly support the idea that Aβ is
a drop, what we don’t know is an ocean”? permit rapid ex vivo analysis of prion amplifi- the disease-causing entity1. In mouse mod-
cation and the contribution of other cell types els of Alzheimer’s disease, their limitations
Looking at the amount of literature published to amplification, as well as screening of com- notwithstanding, therapeutic approaches that
on one topic related to neurodegeneration (for pounds that might protect against neurotoxic- target Aβ reduce disease-specific pathology
instance, the generation of amyloid-β (Aβ) ity9. Thus, the prion field has a set of tools that and ameliorate memory deficits13,14. The
peptide), one might be surprised to read such allow the dissection of pathological cascades field, however, tends to place undue empha-
a skeptical impression. The identification of downstream of PrPsc and the identification of sis on oligomeric Aβ as the disease-causing
the proteolytic machinery involved in Aβ targets to stem brain damage. entity to explain the discrepancies described
generation1 paved the way for amyloid-based Researchers investigating Alzheimer’s dis- above. Do we know the precise biochemical
therapeutic strategies. We know a number of ease lack such models and, at best, have models or structural characteristics and direct func-
disease-causing genes and risk factors, and that can recapitulate mild cognitive impair- tional effects of a disease-initiating oligomer?
research on neurodegenerative disease has also ment (MCI) and the pathology of Alzheimer’s Amyloid oligomers vary in size and shape
provided unexpected discoveries of signaling disease but not true neurodegeneration. As an from small soluble oligomers such as dimers
and protein degradation pathways linked to Alzheimer’s researcher, I strongly support the and trimers, to multimers (such as Aβ*56)
autophagy2, neurogenesis3, myelination4–6 and amyloid cascade hypothesis10 (Fig. 1) but feel and other assemblies, including Aβ-derived
regulated intramembrane proteolysis7. Many we have not obtained definitive proof that Aβ is diffusible ligands, annular complexes and
important questions, however, remain unan- the causative agent of neurodegeneration, nor protofibrils15. Oligomers and fibers are in
swered. Chief among them are the proximal have we conclusively identified what forms of equilibrium between aggregation and disag-
causes of neurodegeneration and how a more Aβ initiate neurodegeneration (Fig. 1). The gregation, so it is unlikely that one specific
holistic view of these triggers inform future pathologic characteristics of Alzheimer’s dis- oligomeric species causes neuronal loss. It
therapeutic strategies. ease (namely plaques and tangles) have been seems more likely that many types of soluble
described for over 100 years, and extensive oligomers exert toxicity.
What is the nature of the neurotoxic biochemical and cell biology studies have con- Small oligomers have been purified from
amyloid species? firmed that both Aβ and tau are neurotoxic. the conditioned medium of cells16 (although
The most relevant question is as follows: what Yet, plaques are also found in cognitively nor- here preferentially one cell line is issued17)
mal individuals, and plaque burden does not and from human brains18,19. But do soluble
1DZNE—German Center for Neurodegenerative correlate with memory decline. A recent study oligomers really exist in vivo, or are they gen-
Diseases, Munich, Germany. 2Adolf-Butenandt- in a cohort of cognitively intact nuns confirms erated during the isolation protocol, which in
Institute, Biochemistry, Ludwig-Maximilians- these observations and posits that compensa- all cases includes procedures during which
University, Munich, Germany. tory mechanisms emerge in the face of exten- isolated molecules are concentrated (a step
e-mail: chaass@med.uni-muenchen.de sive amyloid burden11, or, more provocatively, that in itself could initiate oligomerization)?
calls into question the amyloid cascade hypoth- It is hard to characterize the in vivo properties
Published online 21 September 2010; esis. Moreover, successful removal of amyloid of oligomers, and new, highly sensitive bio-
doi: 10.1038/nm.2223 plaques by immunotherapy fails to improve physical technologies would be required to
nature medicine advance online publication 1
commentary
visualize them directly in vivo. Furthermore, Changes in Aβ metabolism
Increase in total Aβ
imagine that a homogenous Aβ oligomer Increase in the Aβ42/Aβ40 ratio
is isolated that has defined physical and Reduced degradation
neurotoxic characteristics and is applied to pri-
mary neurons, leading to neurotoxicity and in
vivo, to a reduction in long-term potentiation Oligomerization of Aβ42 and diffuse plaque deposits
and memory deficits. Do these experiments
What is the nature of the oligomeric species?
provide evidence of a disease-causing entity? Is there a selective Aβ receptor?
It remains unclear whether these aggregates
Subtle effects of soluble oligomers on synapse function
are still of the same size and structure in vivo,
or whether they continue to aggregate and Is LTP a good readout?
are modified by processes such as oxidation
and cross-linking (Fig. 1). Another important Abnormal distribution of tau and aberrant tau phosphorylation and dephosphorylation
issue is whether long-term potentiation is the
best readout for these experiments, as it seems How is Aβ linked to tau?
to be extremely sensitive to many kinds of cel-
lular manipulations. Moreover, these oligom- Oligomerization of tau
ers should not only affect synaptic function What is the nature of the oligomeric species?
but also contribute to the massive neuronal
loss observed in individuals with Alzheimer’s Propagation and neuronal dysfunction leading to cell death
disease. Although oligomers are apparently
present in mouse models, only limited neu- What is required for propagation?
ronal loss is seen in these models. Why are certain neurons resistant to cell death?
Which signaling pathways lead to cell death?
This also raises the question of the specific
Dementia with
downstream effects of Aβ toxicity. Do these plaque and tangle pathology
oligomeric Aβ species bind specific receptors?
What is the role of plaques and tangles?
Although one such receptor could be cellu- How do environmental factors and aging affect
lar PrP (PrPc)20, studies of PrPc-knockout the development of pathology and dementia?
mice have produced conflicting results21,22.
Alternatively, do the toxic effects of Aβ rely Figure 1 The amyloid cascade hypothesis. A number of questions need to be addressed to
on nonspecific interactions with other pro- understand this pathogenic cascade and the link between Aβ and tau pathology. LTP, long-term
teins or even lipid membranes (Fig. 1)? potentiation.
Another point that is not yet fully under-
stood is the reliance of Aβ-dependent toxic-
ity on the presence of tau. Inhibition of tau evidence that neuronal cell death can occur impetus for additional trials of Aβ-directed
expression blocks seizures induced by the independently of and even before tangle therapeutic agents in people with sporadic
Aβ-mediated overstimulation of excitatory formation26,27. Oligomers of tau might be Alzheimer’s disease.
N-methyl-d-aspartate (NMDA) receptors involved in disease processes, but the physi-
and improves survival in a transgenic mouse cal nature of such putative oligomers remains A unifying mechanism of disease
model of Alzheimer’s disease23. In that regard, to be elucidated (Fig. 1). propagation
it is interesting that Aβ toxicity, which is An alternative approach to test the amyloid
largely thought to occur on the postsynaptic A preventive trial to prove the amyloid cascade hypothesis is to determine whether
membrane, has been shown to be meditated cascade hypothesis disease-causing agents are able to induce
by the axonal tau protein24. Surprisingly, tau Ultimately, a proof-of-principle clinical spreading or propagation of the disease. What
targets Fyn kinase to postsynaptic densities, trial of a therapeutic strategy that lowers Aβ types of Aβ species are sufficient to promote
where it phosphorylates and stabilizes the abundance would provide clear evidence in further aggregation of native proteins and
NMDA receptor subunit 2. Trapping tau in support of the amyloid cascade hypothesis. initiate a neurodegenerative cascade? Studies
the soma prevents phosphorylation of NMDA Recent clinical trials of Aβ immunotherapy by Mathias Jucker and his colleagues dem-
receptor subunit 2 and blocks excitotoxic- failed to show much cognitive improvement onstrated that brain homogenates contain-
ity. Of note, the earliest pathological change despite a reduction in amyloid burden12. The ing Aβ were sufficient to induce Alzheimer’s
observed during the course of Alzheimer’s clinical symptoms of Alzheimer’s disease are disease–like pathology28. Moreover, patho-
disease is the redistribution of tau from the thought to present after a long prodromal logical aggregates of α-synuclein and tau
axon to the somatodendritic compartment25. phase, during which considerable neuronal can also ‘infect’ neighboring cells and induce
In Alzheimer’s disease, enhanced amounts of loss occurs. Ideally, one would need to treat pathology reminiscent of Parkinson’s dis-
Fyn might reach dendritic spines and enhance patients with a defined risk (such as carriers of ease and frontotemporal dementia, respec-
Aβ-mediated toxicity. As these events appar- presenilin mutations) presymptomatically in tively29–33. Adriano Aguzzi hypothesizes
ently occur before neurofibrillary tangles are a small preventive trial. If patients who were that many if not all amyloid proteins such as
formed, this calls into question the relevance treated presymptomatically with an anti-Aβ Aβ, tau, α-synuclein, amyloid A and poly-
of these large intracellular inclusions in neu- vaccine or secretase inhibitor or modulator glutamine proteins are “prionoids” capable
ronal cell death (Fig. 1). Recent studies are showed improved cognition or a delay in of amplifying themselves via conformational
consistent with this notion and have provided the onset of disease, this could provide the alterations34. Nevertheless, data from prion
2 advance online publication nature medicine
commentary
disease research suggests that spreading and be identified, new therapeutic agents can be the mutant protein, might determine whether
neurotoxic signaling may not necessarily be targeted to specific nodes within these net- a given anatomical region or cell type is resis-
linked35. Moreover, do these prionoids act works. Caution is warranted, as systems biol- tant or vulnerable to the toxic effects of the
alone? Probably not, as injection of synthetic ogy approaches have proven most useful in protein.
Aβ preparations, including purified oligom- analyzing simple model organisms and are only
ers thought to be the neurotoxic entity (see beginning to be applied to disease-oriented Environmental influences—one hit is not
above), fails to induce spreading, suggesting biomedical research in higher organisms. enough
additional cofactors may be needed28. Owing to recent advances in genomics There are many diseases in which the age of
and proteomics, comprehensive quantita- onset of carriers with a specific autosomal
Selective vulnerability and focal tive approaches can be applied to smaller and dominant mutation varies substantially, sug-
induction of disease propagation smaller amounts of material, making an inte- gesting that there are additional risk factors.
Neuronal degeneration spreads in a stereotyped grative investigation of disease tissue possible. Recent findings support a two-hit hypothesis
fashion, with certain regions of the brain (such The emergence of bioimaging and new models for some genetically inherited cases of amyo-
as the cerebellum) being spared until the late of neurodegenerative disease allow a dynamic trophic lateral sclerosis47. For example, muta-
stages of Alzheimer’s disease36. Why are certain assessment of cellular and molecular interac- tions in fused in sarcoma (FUS) cause this
populations of neurons selectively vulnerable tions from the meso- to the nanoscale. One form of the disease48,49. These mutations lead
to apoptosis while others remain resistant (Fig. caveat is that unbiased research strategies must to the redistribution of the protein from the
1)? This selective vulnerability might result in always be based on solid, functional readouts, nucleus to the cytosol where, upon addition of
part from differences in the lipid composition even on single genes and proteins. stressors, the protein forms aggregates charac-
of neuronal membranes. Pathological Aβ gen- Without that detailed knowledge, holistic teristic of stress granules47. Long-term stress
eration by secretases is strongly affected by the approaches might generate complex interaction may precipitate formation of large deposits by
surrounding lipid composition37. Alternatively, networks that in the end may have little if any fusion of stress granules and cause disease. As
we might gain some insight from cells that are relevance in vivo. The identification of hundreds expression of the mutant protein alone does
seemingly resistant to neurodegeneration, such of protein-protein interactions will not help us not result in aggregate formation, these results
as cerebellar neurons in Alzheimer’s disease36. to understand neurodegeneration unless we suggest that two events are needed: a gain- or
Systems biology approaches including genom- investigate their functional importance at the loss-of-function mutation in a protein, leading
ics, proteomics and lipidomics could be used cellular level. In other words, we should avoid to its altered cellular function or distribution,
to identify alterations in proteins or in the lipid producing data instead of knowledge. and an additional stressor to promote aggre-
composition of these cells that confer vulner- gate formation. Environmental factors greatly
ability or resistance to the toxic effects of Aβ. Altered proteolysis affect disease onset in Parkinson’s disease50,
One process that might be dysregulated in and environmental enrichment reduces the
An integrated picture of disease protein-folding disorders such as Alzheimer’s, cognitive deficits observed in a transgenic
pathogenesis Parkinson’s and Huntington’s diseases is pro- mouse model of Alzheimer’s disease51. In
An integrative approach to the analysis of dis- tein degradation2. Inhibition of the ubiquitin- fact, clinicians sometimes suggest physi-
ease will aid in obtaining a full picture of the proteasome system is sufficient to induce neu- cal and mental exercise to delay the onset of
pathology of Alzheimer’s disease, which not ronal death39, and altered proteasome function Alzheimer’s disease. A recent study showed
only consists of amyloid plaques and neuro- has been observed in Alzheimer’s disease40. A that Aβ generation is reduced during sleep and
fibrillary tangles but also incorporates cere- number of proteins that have key roles in neu- increases during phases of activity52. Are these
bral amyloid angiopathy and inflammatory rodegeneration, such as huntingtin, androgen two findings counterintuitive? Perhaps not, as
responses. Traditionally, neurological diseases receptor, ataxin-3, tau and α-synuclein, are de novo Aβ production (as monitored in the
have been separated into mechanistically cleared by autophagy2. Autophagic clearance sleep study) and Aβ clearance (as observed in
distinct families such as neurodegenerative, of small aggregates is affected by aging and the environmental enrichment studies) may be
inflammatory or vascular conditions. This altered in disease41,42, and recent findings carefully balanced in vivo, and slight changes
classification was predicated on the notion suggest that presenilin mutations affect this in either pathway could greatly affect disease
that clinical phenotypes relate in a categorical proteolytic pathway43. Are aging-associated pathogenesis.
fashion to a discernable disease mechanism. alterations in autophagy an underlying cause
As a result, research efforts have traditionally of neurodegeneration? Translation, that’s what matters
reflected this categorization and tend to focus Alterations in proteolysis would be pre- It is clear that although substantial progress
on one or another of these mechanisms in iso- dicted to initiate degeneration simultaneously has been made in understanding the cellular
lation, with little or no crosstalk. in many brain regions, but these diseases are mechanisms of neurodegeneration, problems
Yet, there is a clear interaction between associated with a regional focus in terms of arise as soon as these findings are translated
neurodegeneration, vascular dysfunction and disease initiation. For example, in individu- into the clinic. In other words, we are ready
inflammation. For example, elimination of als with frontotemporal lobar degeneration, to treat Alzheimer’s disease–like pathology
microglia severely increases prion titers9, and autosomal dominant mutations in the pro- and probably related memory phenotypes in
microglia are involved in the removal of amy- granulin gene44,45 lead to the aggregation and animal models, but there are major hurdles to
loid plaques after vaccination against Aβ38. In ubiquitous deposition of the TAR DNA bind- the treatment of patients. As discussed above,
addition, analysis of several neurodegenerative ing protein-43 in the brain. Imaging studies on we need to be able to identify individuals at
disorders using systems biology approaches affected individuals, however, reveal that the risk for neurodegeneration as early as pos-
might reveal that an intricate network of shared disease does not start evenly in all parts of the sible (before profound neuronal cell loss). To
mechanisms initiates the neurodegenerative brain but begins with asymmetric atrophy46. this end, new biomarkers and more sensitive
cascade. If shared mechanisms of disease can This suggests that other factors, in addition to in vivo imaging techniques that can differ-
nature medicine advance online publication 3
commentary
entiate disease-causing oligomeric species 7. Brown, M.S., Ye, J., Rawson, R.B. & Goldstein, J.L. 284, 12845–12852 (2009).
Cell 100, 391–398 (2000). 30. Clavaguera, F. et al. Nat. Cell Biol. 11, 909–913
from the large and potentially inert protein 8. Aguzzi, A. & Haass, C. Science 302, 814–818 (2009).
deposits are required. (2003). 31. Kordower, J.H., Chu, Y., Hauser, R.A., Freeman, T.B. &
9. Falsig, J. et al. Nat. Neurosci. 11, 109–117 (2008). Olanow, C.W. Nat. Med. 14, 504–506 (2008).
For those perplexed by the complexity of the 32. Li, J.Y. et al. Nat. Med. 14, 501–503 (2008).
10. Hardy, J. & Selkoe, D.J. Science 297, 353–356
problem at hand, Sir Isaac Newton provides us (2002). 33. Aguzzi, A. & Rajendran, L. Neuron 64, 783–790
with some more guidance: “nature is pleased 11. Iacono, D. et al. Neurology 73, 665–673 (2009). (2009).
12. Holmes, C. et al. Lancet 372, 216–223 (2008). 34. Aguzzi, A. Nature 459, 924–925 (2009).
with simplicity.” Moving forward, perhaps this 35. Chesebro, B. et al. Science 308, 1435–1439 (2005).
13. Schenk, D. et al. Nature 400, 173–177 (1999).
quote can provide a guiding principle for neu- 14. Schenk, D., Hagen, M. & Seubert, P. Curr. Opin. 36. Thal, D.R., Capetillo-Zarate, E., Del Tredici, K. & Braak,
H. Sci. SAGE KE 2006, re1 (2006).
rodegenerative research. Immunol. 16, 599–606 (2004).
37. Fraering, P.C. et al. Biochemistry 43, 9774–9789
15. Haass, C. & Selkoe, D.J. Nat. Rev. Mol. Cell Biol. 8,
(2004).
ACKNOWLEDGMENTS 101–112 (2007).
38. Bard, F. et al. Nat. Med. 6, 916–919 (2000).
This article reports on some of the main points 16. Walsh, D.M. et al. Nature 416, 535–539 (2002).
39. Winklhofer, K.F., Tatzelt, J. & Haass, C. EMBO J. 27,
17. Podlisny, M.B. et al. Biochemistry 37, 3602–3611
raised at the Herrenhausen Symposium on 336–349 (2008).
(1998).
Neurodegeneration in Seeon, Germany (May 2010) 40. Keller, J.N., Hanni, K.B. & Markesbery, W.R. J.
18. Lesné, S. et al. Nature 440, 352–357 (2006).
during the session on the initiation and propagation Neurochem. 75, 436–439 (2000).
19. Shankar, G.M. et al. Nat. Med. 14, 837–842 (2008).
41. Ravikumar, B., Duden, R. & Rubinsztein, D.C. Hum.
of neurodegeneration. I thank A. Aguzzi, 20. Laurén, J., Gimbel, D.A., Nygaard, H.B., Gilbert, J.W. & Mol. Genet. 11, 1107–1117 (2002).
D. Rubinsztein, S. Sisodia, D. Edbauer and M. Meyer- Strittmatter, S.M. Nature 457, 1128–1132 (2009). 42. Cataldo, A.M., Hamilton, D.J., Barnett, J.L., Paskevich,
Lühmann for reading this manuscript. This work was 21. Calella, A.M. et al. EMBO Mol. Med. 2, 306–314 P.A. & Nixon, R.A. J. Neurosci. 16, 186–199 (1996).
supported by the Deutsche Forschungsgemeinschaft. (2010). 43. Lee, J.H. et al. Cell 141, 1146–1158 (2010).
22. Gimbel, D.A. et al. J. Neurosci. 30, 6367–6374 44. Baker, M. et al. Nature 442, 916–919 (2006).
COMPETING FINANCIAL INTERESTS (2010). 45. Cruts, M. et al. Nature 442, 920–924 (2006).
23. Roberson, E.D. et al. Science 316, 750–754 (2007). 46. Rohrer, J.D. et al. Neuroimage published online,
The author declares no competing financial interests.
24. Ittner, L.M. et al. Cell 142, 387–397 (2010). doi:10.1016/j.neuroimage.2009.12.088 (4 January
25. Ballatore, C., Lee, V.M. & Trojanowski, J.Q. Nat. Rev. 2010).
1. Haass, C. EMBO J. 23, 483–488 (2004). Neurosci. 8, 663–672 (2007). 47. Dormann, D. et al. EMBO J. 29, 2841–2857 (2010).
2. Rubinsztein, D.C. Nature 443, 780–786 (2006). 26. de Calignon, A. et al. Nature 464, 1201–1204 48. Kwiatkowski, T.J. Jr. et al. Science 323, 1205–1208
3. Veeraraghavalu, K., Choi, S.H., Zhang, X. & Sisodia, (2010). (2009).
S.S. J. Neurosci. 30, 6903–6915 (2010). 27. Paquet, D. et al. J. Clin. Invest. 119, 1382–1395 49. Vance, C. et al. Science 323, 1208–1211 (2009).
4. Hu, X. et al. Nat. Neurosci. 9, 1520–1525 (2006). (2009). 50. Di Monte, D.A., Lavasani, M. & Manning-Bog, A.B.
5. Willem, M. et al. Science 314, 664–666 (2006). 28. Meyer-Luehmann, M. et al. Science 313, 1781–1784 Neurotoxicology 23, 487–502 (2002).
6. Bremer, J. et al. Nat. Neurosci. 13, 310–318 (2006). 51. Lazarov, O. et al. Cell 120, 701–713 (2005).
(2010). 29. Frost, B., Jacks, R.L. & Diamond, M.I. J. Biol. Chem. 52. Kang, J.E. et al. Science 326, 1005–1007 (2009).
4 advance online publication nature medicine
C O M M E N TA RY
Degeneration and repair in central
nervous system disease
Eng H Lo
Divergent disease triggers in disease in neurodegeneration may also act as responses to injury and disease in any organ6.
neurodegeneration may induce stimuli that induce endogenous compensa- The same might be true in the context of neu-
convergent endogenous pathways in tion and recovery. Hence, investigations into rodegeneration.
neuronal, glial and vascular elements the pathophysiology of CNS disease should In spite of the highly divergent disease trig-
as the central nervous system (CNS) take into account not only the primary mecha- gers in neurodegeneration, is it possible that
attempts to compensate, remodel and nisms of degeneration but also simultaneous these diseases also induce convergent down-
recover. Dissecting these multicellular processes of regeneration as the brain tries to stream mechanisms of compensation, repair
mechanisms and the integrative repair itself. and remodeling (Fig. 1a)? This is obviously a
responses in cerebral blood flow speculative idea. But some evidence of endog-
and metabolism may allow us to Divergence in disease, convergence in enous recovery during neurodegeneration can
understand the balance between injury repair? indeed be detected in animal models and clini-
and repair, validate new targets and The mammalian nervous system is highly com- cal studies.
define therapeutic time windows for plex. Many things can go wrong in many ways. It is well known that behavioral and motor
neurodegeneration. Clinically, there are multiple ‘types’ of neuro- adaptation allows for some individuals with
degeneration, comprising major diseases such Parkinson’s disease to compensate during
Remarkable advances have been made in the as Alzheimer’s disease, Parkinson’s disease, the initial stages of disease. But beyond com-
last decade in understanding the basic mecha- amyotrophic lateral sclerosis (ALS), multiple pensation, some degree of actual neuronal
nisms of neurodegeneration. Progress has sclerosis, Huntington’s disease and prion dis- remodeling may also take place. Dendritic
come on many fronts, including the molecular ease. This wide spectrum of neurodegenera- sprouting and reafferentation of damaged
biology of cell death, animal models, genetics tive phenotypes reflects key differences in the areas are known to occur in animal models of
and neuroimaging of clinical disease. Many tar- proximal triggers and pathologic markers of focal brain lesions. The most common animal
gets are being tested in a wide range of experi- disease: plaques and tangles in Alzheimer’s models of Parkinson’s disease involve selective
mental studies and clinical trials. Along with disease, Lewy bodies in Parkinson’s disease, lesioning of dopaminergic neurons within the
the excitement, however, some caution may motoneuron death in ALS and demyelination substantia nigra. Because the substantia nigra
be warranted. Advances in our knowledge of in multiple sclerosis. However, these seemingly projects to the striatum, this depletes striatal
excitotoxicity, oxidative stress, mitochondrial divergent mechanisms also lead to multiple dopamine and mimics clinical Parkinson’s dis-
dysfunction and apoptosis similarly provided convergent pathways. Regardless of the initial ease. However, it is now recognized that local
a rich repertoire of targets and drugs for ‘acute triggers, many overlapping downstream and/ sources of dopamine may also exist in the mam-
neurodegeneration’ after stroke and traumatic or secondary pathways are induced, including malian striatum, deriving in part from intrinsic
brain injury1,2. Yet, almost all clinical trials in neuroinflammation3. An important challenge tyrosine hydroxylase–positive neurons7. After
stroke and brain trauma have failed thus far. in designing therapies for neurodegeneration is loss of dopaminergic input from the substan-
Broadly speaking, there is still no clinically to not only eliminate the initial disease triggers tia nigra, local tyrosine hydroxylase–positive
validated neuroprotectant. This article reports but also ameliorate the deleterious inflamma- neurons seem to remodel and expand in rodent
on one speculative hypothesis: triggers of tory responses within the CNS once the disease and primate models of Parkinson’s disease8,9.
is under way. It has been proposed that similar substrates
Eng H. Lo is at the Neuroprotection Research Although the CNS was originally thought of neuroplasticity may also exist in individu-
Laboratory, Departments of Neurology and to be an immune-privileged organ, it is now als with Parkinson’s disease10. Whether these
Radiology, Massachusetts General Hospital and known that inflammation can be triggered in pathways truly restore clinically relevant dop-
Harvard Medical School, Boston, Massachusetts, injured brain tissue. For example, activation amine function remains to be determined11.
USA. of microglia and responses in both innate and Similar substrates of repair and remodel-
e-mail: lo@helix.mgh.harvard.edu adaptive immunity accompany almost all types ing may also be present in Alzheimer’s dis-
of neurodegeneration4,5. From an evolutionary ease. Indeed, one of the early observations in
Published online 21 September 2010; perspective, inflammation can be interpreted Alzheimer’s disease was that hippocampal
doi: 10.1038/nm.2226 as part of a highly conserved set of endogenous degeneration is often accompanied by a reor-
nature medicine advance online publication 1
commentary
ganization of acetylcholinesterase staining pat- a Cofactors (genetic, environmental,
terns, indicative of cholinergic sprouting from physiological, pharmacological)
basal forebrain regions12. More recent stud-
ies now propose that increased expression of
synaptic proteins, such as postsynaptic density Endogenous response Prolonged inflammation
Initial disease triggers
protein-95, may underlie plasticity in prefrontal to injury and degeneration
and frontal networks as the brain attempts to
cognitively compensate for degenerating neu-
Non–cell-autonomous crosstalk
rons13. Functional magnetic resonance imaging in neuronal, glial
studies suggest that remapping of cortical net- and vascular compartments
works may extend to motor systems as well14. b
Indeed, the degree of cognitive decline may be
Cumulative injury
more closely related to an individual’s endog- Endogenous compensation
Level of response
enous neuronal plasticity than to the cerebral and remodeling
abundance of amyloid or tangles per se15. Recent
work is beginning to elucidate the underlying
mechanisms. Perturbations in amyloid pre-
cursor protein processing lead to increased
amyloid-β generation, assembly of toxic oligom-
ers, plaque formation and Alzheimer’s disease
pathology16. However, homeostatic forms and Time after disease initiation
levels of amyloid-β may also act as functional Optimal treatment
regulators of transmitter release and synaptic window
function17,18. Ultimately, the balance between
beneficial-adaptive and aberrant-maladaptive
Figure 1 Endogenous responses, cofactors and therapeutic time windows in neurodegeneration. The
forms of synaptic and neuronal remodeling wide spectrum of pathology in neurodegeneration reflects a multitude of initial triggers involved in
may significantly influence how Alzheimer’s the induction of disease. But divergent upstream mechanisms also lead to convergent downstream
disease pathology disrupts cerebral function as pathways as the brain responds to disease and injury. Endogenous responses of the CNS may comprise
the disease progresses19. both deleterious and potentially beneficial mechanisms of inflammation and neurovascular remodeling.
Endogenous mechanisms of brain repair Non–cell-autonomous crosstalk between neuronal, glial and vascular compartments form the basis
and recovery can be classified into several cat- for these phenomena, and many cofactors influence the signals and substrates of these endogenous
responses over time. Ultimately, prolonged inflammation and injury lead to a cumulative disease
egories: behavioral compensation, activation
burden that outstrips any endogenous attempts at compensation or remodeling. Understanding
of latent or parallel circuits, neuronal plastic- these transitions may help define therapeutic time windows where candidate treatments for
ity, remodeling of discrete networks and per- neurodegeneration should be more effective.
haps even neurogenesis. Traditionally, it was
thought that no new neurons were generated linked to vascular signaling as part of an evolu- inflammatory cascade. To some degree, then,
after birth in mammalian brains. However, tionarily conserved phenomenon1,2,23. Hence, angiogenesis in diseased brains may represent
a wealth of studies have led to a paradigm neuroplasticity may occur only within the con- a nonspecific epiphenomenon associated with
shift. It is now generally accepted that pock- text of brain microvessel remodeling24. New neurodegeneration.
ets of ongoing neurogenesis persist in the cells and networks need new blood supplies.
adult brain, including active loci in the sub- If endogenous repair and neuronal recovery Multiple mechanisms in multiple cells
ventricular zone lining the lateral ventricles take place in neurodegeneration, is there a The neurovascular unit is an increasingly
and subgranular zone of the dentate gyrus. corresponding angiogenic response as well? accepted conceptual model in neuroscience, in
Emerging data suggest that neurogenesis is Although the literature is sparse, there have which cell-to-cell signaling between neuronal,
surprisingly plastic, and increased neurogen- been a few descriptions of augmented vascular glial and vascular elements underlies both
esis can be observed in models of CNS injury growth in a wide spectrum of CNS diseases, physiology and pathophysiology in the brain1,2.
and disease20. Several suspected molecular including Alzheimer’s disease, Parkinson’s The term ‘neurodegeneration’ implies a purely
mediators in Alzheimer’s disease, such as disease and multiple sclerosis25–29. Insofar neuronal phenomenon, but this is clearly not
amyloid, presenilin 1, Notch and ErbB4, are as triggers of disease may conversely act as the case when one examines the signals and
known to participate in the molecular regula- stimuli for repair, it might be interesting to substrates of disease more closely. Disruptions
tion of neurogenesis21. A survey of the animal ask whether mediators of neurodegeneration in cell-cell signaling within the neurovascu-
model literature reveals that neurogenesis can can promote vascular recovery. For example, lar unit may underlie non–cell-autonomous
sometimes be affected in Alzheimer’s disease, amyloid peptides may augment angiogen- mechanisms during the response to injury and
depending on the stage of disease and the esis by amplifying fibroblast growth factor disease in the CNS32 (Fig. 1a).
underlying phenotypes of the mutant mice signaling30. Furthermore, crosstalk between For example, interactions between neurons
involved22. Whether alterations in neurogen- angiogenesis and neurogenesis may occur, and astrocytes are essential for the regulation
esis actually occur in human neurodegenera- as amyloid has been shown to increase rates of glutamate transmission. Without astrocytes,
tion remains unclear and provides an exciting of neuronal differentiation of bone marrow– neurons become increasingly vulnerable to
frontier for research. derived endothelial precursor cells31. However, excitotoxicity33. Any disruption in astrocytic
An emerging concept in neuroscience sug- it is important to acknowledge that vascular function can markedly promote neurode-
gests that neuronal responses are intricately remodeling is a crucial component of any generation. Transgenic Huntington’s disease
2 advance online publication nature medicine
commentary
model mice that express mutant huntingtin neural repair50,51. The pathophysiology of with overlaps between vascular dementia and
only in cortical pyramidal neurons do not show neurodegeneration will surely include analo- Alzheimer’s disease being especially impor-
neurodegeneration34, whereas mice that express gous interactions between neuronal, glial and tant57. Various Alzheimer’s disease trans-
huntingtin in astrocytes develop age-dependent vascular cells. genic mice could be examined in the context
neurological worsening35. In a mouse model Taken together, non–cell-autonomous of hypertension, diabetes and other forms of
of Niemann-Pick disease, expression of wild- mechanisms in neurodegeneration may pro- vascular dysfunction. Given a body of literature
type Niemann-Pick disease type C1 pro- vide a conceptual framework for asking further on the effects of statins on Alzheimer’s disease,
tein in astrocytes was sufficient to decrease questions. How do these multicell pathways these models could also be used to examine
the rate of neurodegeneration and increase mediate the convergent mechanisms between how cardiovascular medications such as statins
life span36. Primary astrocyte and micro- degeneration and regeneration? Can differences and antihypertensive drugs interact with the
glial cultures derived from the superoxide in cell-to-cell signaling help explain the selec- various pathways targeted in neurodegenera-
dismutase–mutant mouse model of ALS pro- tive neuronal vulnerability that is the signature tion mouse models. Finally, as discussed above,
duce neurotoxic mediators in conditioned phenotype of so many CNS diseases? What are differences in systemic inflammation may be
media that kill wild-type motoneurons37. In the mediators that regulate these processes? crucial for disease pathogenesis. Almost all
mouse models of Alzheimer’s disease, wide- It has been proposed that brain injury and mouse models are developed and assessed in
spread perturbations of calcium signaling were repair sometimes include biphasic mediators rigorously controlled housing conditions. But
detected in astrocyte networks long distances and mechanisms52. The same molecule or cell does this also mean that most of our trans-
away from amyloid plaques38. Hence, protect- can behave differently at different phases of the genic mice have highly ‘clean’ inflammatory
ing neurons alone may not be enough, and disease. Overactivation of N-methyl-d-aspartic baselines, unlike those of a typical human
strategies to improve glial function may also acid (NMDA) receptors induces excitotoxicity, with metabolic and vascular diseases? How do
be important for CNS disease therapies. but without NMDA signaling, neuronal plas- altered inflammatory baselines affect the bal-
Signaling between endothelial cells and ticity cannot take place53. The stress-activated ance between degeneration and regeneration?
neurons may have especially crucial roles in protein kinase c-Jun N-terminal kinase pro- Beyond promoting the risk of disease, these
neurodegeneration23,29,39. Endothelial dys- motes neuronal apoptosis but is also required cofactors may also contribute to resistance to
function and perturbations in microvascular for axonal remodeling54. Activated microglia disease. Research efforts are largely focused on
flow regulation precede the development of are key players in neuroinflammation, but cer- identifying factors and mediators that increase
neuronal dysfunction in some mouse models tain forms of microglia can secrete beneficial the rate of neurodegeneration. But individu-
of Alzheimer’s disease40. Subtle disruptions in neurotrophic factors55. Aggregated proteins als, even with similar loads of disease triggers,
the blood-CNS barrier may be detected before may represent misfolded pathologic species can have widely varying rates of neurological
neuronal death in the superoxide dismutase– or underlie protective endogenous attempts decline. Is it possible that differences in the
mutant mouse model of ALS41,42. Compromised to sequester toxic molecules56. Understanding ability to adapt, repair and remodel partly
blood flow in white-matter regions may precede how these biphasic mechanisms are coordi- underlie this phenomenon? And, if so, might it
lesion development in some individuals with nated in multiple cell types will be essential be productive to screen for genes and cofactors
multiple sclerosis43. Microvessels in the brain for dissecting the precarious balance between that promote compensation and recovery and
might not be simply an inert system for blood neurodegeneration and ongoing compensation enhance resistance to neurodegeneration?
flow. Instead, the cerebral endothelium may and repair.
also serve as an endocrine organ that actively Time is brain
exchanges signaling mediators and trophic fac- Cofactors in risk and recovery The mantra in stroke and trauma clinical tri-
tors with gray and white matter44–46. Disease Advances in genetics have revealed a host of als is “time is brain.” After the initial ischemic,
triggers such as amyloid may disrupt these underlying mechanisms that increase risk of hemorrhagic or traumatic insult, brain cells
forms of trophic coupling between the vascular disease. In neurodegeneration, the power of inexorably begin to die. The longer one waits,
and neuronal compartments. genetics is clearly indicated in the tremendous the more cells are lost, and the less effective
If non–cell-autonomous signaling between number of sophisticated and targeted transgenic any putative neuroprotective therapy might
neurons, glia and blood vessels contributes to mouse models that are now available. However, be. Clinical trial design in stroke and trauma
disease progression, is it possible that com- it might still be useful to ask whether these mice is highly focused on time of treatment, where
pensation and repair also involve coordinated represent models of disease or, more accurately, only patients admitted within specific times
remodeling in the entire neurovascular unit models of mechanisms. These discussions may after onset are enrolled. Is it possible that simi-
(Fig. 1a)? Again, examples can be drawn from not be purely semantic. The initial molecular lar attention to treatment time windows is also
findings in acute brain injury. Angiogenic and triggers of neurodegeneration are cellular, but essential for neurodegeneration (Fig. 1b)?
neurogenic plasticity are tightly coregulated pathophysiology is expressed at the organ level, If one accepts the proposed hypothesis of
after stroke and brain trauma47. This might and clinical disease is usually manifested in ongoing degeneration and regeneration in
not be surprising, as molecular mechanisms of patients with numerous other health problems CNS disease, then one would predict that ini-
neurogenesis and angiogenesis have been evo- requiring concurrent medications. tially, the disease burden would be light and
lutionarily conserved so that similar mediators Would it be useful to consider the devel- neuronal dysfunction could be ameliorated
and pathways are involved in both phenom- opment of combination approaches whereby by endogenous mechanisms of compensation,
ena48. After experimental stroke in rat mod- such cofactors are incorporated into the mouse remodeling and repair. At this stage, patients
els, newborn neurons are seen to migrate along models (Fig. 1a)? Because aging is a key cofac- might still be asymptomatic or relatively well.
‘highways’ that seem to run in close proximity tor in neurodegeneration, future studies should As the disease continued to progress beyond
to remodeling blood vessels49. Promotion of aim to test leading candidate therapies in aged certain thresholds, the burden of disease would
neural plasticity enhances vascular regrowth; transgenic mice. Concomitant vascular dis- overcome the brain’s endogenous ability to
conversely, angiogenic stimulation enhances ease may be a crucial cofactor in dementia, cope. During this transition phase, neurode-
nature medicine advance online publication 3
commentary
generation would accelerate, and saving the worsening balance between degeneration and for conducting rigorously powered, statistically
brain might become extremely difficult, no regeneration may ultimately allow us to find controlled screening of candidate mechanisms,
matter how potent the therapy may be. Hence, patients who still have a chance (Fig. 1b). targets and drugs. Findings could be replicated
under some conditions, preventing disease in multiple models across multiple laborato-
progression might be more effective than try- Collaborations and consortia ries. The role of cofactors (age, gender, envi-
ing to cure the brain after decades of neuronal Complex problems require collaborative ronment, baseline inflammation, metabolic
dysfunction and death. solutions. To begin with, the basic molecular and cardiovascular disease, and concurrent
An example of this concept of ‘prevention’ mechanisms of neurodegeneration are highly medications) could be more easily assessed.
versus ‘cure’ can be observed in the mouse multifactorial. If the proposed phenomenon Different laboratories could also contribute
model literature for ALS. Effective neuro of simultaneous compensation and repair is expertise in dissecting non–cell-autonomous
protection—that is, decreased motor impair- indeed ongoing, this would add yet another crosstalk between various neuronal, glial and
ment and increased life span—has been reported layer of complexity. Traditional approaches vascular cell types in the mammalian CNS.
for a wide range of targets in superoxide involving a single model, single pathway and Molecular mechanisms of selective neuronal
dismutase–mutant mouse models of ALS58. Yet single laboratory may not be enough. To dissect death may not make sense unless interpreted in
a closer look would reveal that the majority of these underlying mechanisms of degeneration the context of integrated responses in cerebral
these studies involved treatment of mice in the and regeneration in the entire neurovascular blood flow and metabolism.
very early stages of disease. In contrast, most unit, multidisciplinary networks and consortia In spite of divergent disease triggers, it
clinical trials in ALS recruit subjects that are will be useful. is likely that convergent downstream path-
sometimes quite far along in terms of disease The richness of available models in neuro- ways of secondary response and remodeling
progression and severity. This phenomenon is degeneration may be both an advantage and will be activated during neurodegeneration.
by no means unique to ALS. A recent study in a disadvantage. The many cellular and animal Dissecting the interplay between injury and
the TG4510 tau-mutant mouse showed that, models provide diverse and powerful oppor- repair may allow us to find therapies that
during the early stages of disease, many neu- tunities for generating and testing hypotheses maximize neuroprotection and neurorepair.
rons were positive for active caspase but had not at the mechanistic level. However, in terms of Ultimately, understanding these transitions
yet undergone apoptosis, suggesting that tau- serving as preclinical platforms for drug testing, and mechanisms may help us find ways to
bearing neurons are surprisingly long lived59. In they can also pose a challenge. Which models identify patients with compensatory reserves
an inducible cell model of tauopathy, turning off should one use for screening, development and who still have salvageable brains.
the gene encoding mutant tau allowed neurons optimization? Model systems span a wide phy-
ACKNOWLEDGMENTS
to recover before degenerative cascades were logenetic range, comprising individual cells,
The author apologizes to colleagues whose important
too far gone60. Similarly, amyloid-β immuno- Caenorhabditis elegans, Drosophila melano- work could not be directly cited. Because of space
therapy may work better at reducing amyloid gaster, zebrafish, mice and perhaps even non- limitations, mostly review articles were used as
load and deposition as a preventive measure human primates. The workhorse in the field starting points for discussion. Many thanks to
rather than after pathology is established61. remains the transgenic mouse, but the major- B. Bacskai, D. Selkoe, B. Hyman, M. Schwarzschild
and M.M. Ning for helpful discussions; and F. Beal,
Hence, timing is crucial, and there might be ity of these highly targeted mice may represent D. Cleveland, E. Mandelkow, W. Robberecht and
windows of time in which Alzheimer’s disease models of mechanisms rather than standalone all participants in the Herrenhausen Symposium
therapies are more effective. Indeed, the major- models of human disease. Without collabora- on Neurodegeneration for a wonderful educational
ity of experimental treatments for Alzheimer’s tions, the increasing heterogeneity of models experience.
disease have been tested in relatively young and techniques may prove intractable. COMPETING FINANCIAL INTERESTS
mutant mice, whereas clinical trials may typi- A recent meta-analysis of the Alzheimer’s The author declares no competing financial interests.
cally involve a much more heterogeneous pop- disease mouse model literature clearly showed
1. Lo, E.H., Dalkara, T. & Moskowitz, M.A. Nat. Rev.
ulation of dementia patients62. However, it is that many of the models being tested involved Neurosci. 4, 399–415 (2003).
important to recognize that translating time presymptomatic mice62. Perhaps most impor- 2. Moskowitz, M.A., Lo, E.H. & Iadecola, C. Neuron 67,
windows in mice into relevant times for therapy tant, many studies were underpowered and 181–198 (2010).
3. Glass, C.K., Saijo, K., Winner, B., Marchetto, M.C. &
in humans may also be difficult. lacked proper randomization, blinding and Gage, F.H. Cell 140, 918–934 (2010).
Ultimately, early treatments for neurodegen- statistical controls. It is notable that similar 4. Block, M.L. & Hong, J.S. Prog. Neurobiol. 76, 77–98
eration should logically reap greater benefits. conclusions were reached in a meta-analysis of (2005).
5. Lucin, K.M. & Wyss-Coray, T. Neuron 64, 110–122
The sooner one tackles the disease, the better. animal stroke models64. An emerging consen- (2009).
But how would one find these patients? By sus in the stroke field suggests that consortia 6. Medzhitov, R. Nature 454, 428–435 (2008).
7. Betarbet, R. et al. J. Neurosci. 17, 6761–6768
definition, the individuals with earliest-stage are needed to unravel these difficult differences (1997).
disease may be asymptomatic. Neuroimaging between models and laboratories65. Would col- 8. Blanchard, V. et al. J. Neurochem. 64, 1669–1679
or tissue biomarkers may be required63. If sur- laborative consortia also be productive in neu- (1995).
9. Tandé, D. et al. Brain 129, 1194–1200 (2006).
rogate markers (imaging of amyloid, tau or rodegeneration? Early success in the Alzheimer 10. Porritt, M.J. et al. Lancet 356, 44–45 (2000).
neurotransmitter depletion) can be quantified Disease Neuroimaging Initiative suggests that 11. Huot, P., Levesque, M. & Parent, A. Brain 130, 222–
and proven to rigorously correlate with clinical broadly collaborative networks can indeed be 232 (2007).
12. Hyman, B.T., Kromer, L.J. & Van Hoesen, G.W. Ann.
states, then one might envision clinical trials built, where data are shared and findings can be Neurol. 21, 259–267 (1987).
using biomarkers not only for subject selection rapidly disseminated and leveraged. 13. Leuba, G. et al. Neurobiol. Dis. 30, 408–419 (2008).
14. Agosta, F. et al. Hum. Brain Mapp. 31, 515–525
and recruitment but also for the assessment of (2010).
secondary outcome measures. No matter how Questions and opportunities 15. Iacono, D. et al. Neurology 73, 665–673 (2009).
potent one’s proposed target or drug, biologi- Many questions remain, but these translational 16. Selkoe, D.J. Behav. Brain Res. 192, 106–113
(2008).
cal variation will always reveal responders and challenges also present us with new opportuni- 17. Abramov, E. et al. Nat. Neurosci. 12, 1567–1576
nonresponders. Defining the progressively ties. Collaborative approaches should be useful (2009).
4 advance online publication nature medicine
commentary
18. Pearson, H.A. & Peers, C. J. Physiol. (Lond.) 575, 5–10 162–168 (1989). J. Neurosci. 26, 13007–13016 (2006).
(2006). 34. Gu, X. et al. Neuron 46, 433–444 (2005). 51. Taguchi, A. et al. J. Clin. Invest. 114, 330–338
19. Palop, J.J. et al. Neuron 55, 697–711 (2007). 35. Bradford, J. et al. J. Biol. Chem. 285, 10653–10661 (2004).
20. Koch, P., Kokaia, Z., Lindvall, O. & Brustle, O. Lancet (2010). 52. Lo, E.H. Nat. Med. 14, 497–500 (2008).
Neurol. 8, 819–829 (2009). 36. Zhang, M. et al. J. Neurosci. Res. 86, 2848–2856 53. Ikonomidou, C. & Turski, L. Lancet Neurol. 1, 383–386
21. Lazarov, O. & Marr, R.A. Exp. Neurol. 223, 267–281 (2008). (2002).
(2010). 37. Nagai, M. et al. Nat. Neurosci. 10, 615–622 (2007). 54. Waetzig, V., Zhao, Y. & Herdegen, T. Prog. Neurobiol.
22. Marlatt, M.W. & Lucassen, P.J. Curr. Alzheimer Res. 7, 38. Kuchibhotla, K.V., Lattarulo, C.R., Hyman, B.T. & 80, 84–97 (2006).
113–125 (2010). Bacskai, B.J. Science 323, 1211–1215 (2009). 55. Perry, V.H., Nicoll, J.A. & Holmes, C. Nat. Rev. Neurol.
23. Zacchigna, S., Lambrechts, D. & Carmeliet, P. Nat. Rev. 39. Iadecola, C. Nat. Rev. Neurosci. 5, 347–360 (2004). 6, 193–201 (2010).
Neurosci. 9, 169–181 (2008). 40. Iadecola, C. Cell. Mol. Neurobiol. 23, 681–689 56. Williams, A.J. & Paulson, H.L. Trends Neurosci. 31,
24. Madri, J.A. J. Physiol. Pharmacol. 60 Suppl 4, 95–104 (2003). 521–528 (2008).
(2009). 41. Garbuzova-Davis, S. et al. PLoS One 2, e1205 57. Fotuhi, M., Hachinski, V. & Whitehouse, P.J. Nat. Rev.
25. Barcia, C., Emborg, M.E., Hirsch, E.C. & Herrero, M.T. (2007). Neurol. 5, 649–658 (2009).
Front. Biosci. 9, 277–282 (2004). 42. Zhong, Z. et al. Nat. Neurosci. 11, 420–422 (2008). 58. Benatar, M. Neurobiol. Dis. 26, 1–13 (2007).
26. Desai, B.S., Schneider, J.A., Li, J.L., Carvey, P.M. & 43. Varga, A.W. et al. J. Neurol. Sci. 282, 28–33 (2009). 59. de Calignon, A. et al. Nature 464, 1201–1204
Hendey, B. J. Neural Transm. 116, 587–597 (2009). 44. Arai, K. & Lo, E.H. J. Neurosci. 29, 4351–4355 (2010).
27. Holley, J.E., Newcombe, J., Whatmore, J.L. & Gutowski, (2009). 60. Wang, Y., Kruger, U., Mandelkow, E. & Mandelkow, E.M.
N.J. Neurosci. Lett. 470, 65–70 (2010). 45. Dugas, J.C. et al. J. Neurosci. 28, 8294–8305 Neurodegener. Dis. 7, 103–107 (2010).
28. Vagnucci, A.H. Jr. & Li, W.W. Lancet 361, 605–608 (2008). 61. Lemere, C.A. & Masliah, E. Nat. Rev. Neurol. 6, 108–
(2003). 46. Guo, S. et al. Proc. Natl. Acad. Sci. USA 105, 7582– 119 (2010).
29. Zlokovic, B.V. Trends Neurosci. 28, 202–208 (2005). 7587 (2008). 62. Zahs, K.R. & Ashe, K.H. Trends Neurosci. 33, 381–399
30. Cantara, S. et al. FASEB J. 18, 1943–1945 (2004). 47. Arai, K., Jin, G., Navaratna, D. & Lo, E.H. FEBS J. 276, (2010).
31. Jin, H.K., Bae, J.S., Furuya, S. & Carter, J.E. Cell Prolif. 4644–4652 (2009). 63. Hampel, H. et al. Nat. Rev. Drug Discov. 9, 560–574
42, 571–586 (2009). 48. Carmeliet, P. & Tessier-Lavigne, M. Nature 436, 193– (2010).
32. Ilieva, H., Polymenidou, M. & Cleveland, D.W. J. Cell 200 (2005). 64. O’Collins, V.E. et al. Ann. Neurol. 59, 467–477
Biol. 187, 761–772 (2009). 49. Thored, P. et al. Stroke 38, 3032–3039 (2007). (2006).
33. Rosenberg, P.A. & Aizenman, E. Neurosci. Lett. 103, 50. Ohab, J.J., Fleming, S., Blesch, A. & Carmichael, S.T. 65. Fisher, M. et al. Stroke 40, 2244–2250 (2009).
nature medicine advance online publication 5
C O M M E N TA RY
The benefits and limitations of animal
models for translational research in
neurodegenerative diseases
Mathias Jucker
Age-related neurodegenerative tions, such as mutant SOD-linked canine mal models, have driven the development of
diseases are largely limited to humans degenerative myelopathy, resembling amyo- genetically modified animal models based on
and rarely occur spontaneously in trophic lateral sclerosis (ALS))3. familial disease mutations.
animals. Genetically engineered mouse Aged mammals, similar to aged, cognitively
models recapitulate aspects of the normal humans, can develop the neuropatho- Gentically engineered animal models
corresponding human diseases and logical lesions that define human Alzheimer’s Drosophila melanogaster, Caenorhabditis elegans
are instrumental in studying disease disease. For example, cerebral β-amyloidosis and zebrafish have been useful in dissecting
mechanisms and testing therapeutic spontaneously occurs in aged nonhuman basic disease mechanisms and in screening com-
strategies. If considered within the primates, bears and dogs4–7. Neurofibrillary pounds that target such basic mechanisms19,20.
range of their validity, mouse models tangles have been described in aged nonhu- However, the much simpler nervous systems of
have been predictive of clinical man primates, bears and sheep4,8–10. No such nonmammalian species and the species’ phylo-
outcome. Translational failure is less the spontaneously occurring lesions have been genetic distance from humans limit their use as
result of the incomplete nature of the reported in aged laboratory rodents or lower- translational models of human-specific neuro-
models than of inadequate preclinical order species. degenerative diseases.
studies and misinterpretation of the Paradoxically, the paucity of age-related The most popular animal models for study-
models. This commentary summarizes neurodegenerative diseases in nonhuman ing human age-related neurodegenerative
current models and highlights key species could yield some mechanistic insights diseases are models involving genetically engi-
questions we should be asking about into the etiology of the human diseases. At neered mice17,19,21–27. For an updated list of
animal models, as well as questions that least for nonhuman primates, the amino acid current models, see http://www.alzforum.org/
cannot be answered with the current sequences of the disease-defining pathogenic res/com/tra/default.asp. Some general findings
models. proteins are very similar to, or predict an even relevant to translational research with current
more pathogenic protein than, the human mouse models are highlighted below.
Natural animal models sequences11–14. Differences in chaperones, Alzheimer’s disease and cerebral
Age-related neurodegenerative diseases are proteasomal and autophagosomal clearance β-amyloidosis. The discovery that mutations in
largely human-specific diseases. Although mechanisms, lifespan or comorbid conditions the genes encoding amyloid-β precursor protein
aged nonhuman primates and some other are considered causes for the apparent resis- (APP) and presenilins 1 or 2 (PSEN1 or PSEN2)
higher-order animal species show aspects tance of nonhumans to age-related neurode- cause autosomal dominant Alzheimer’s disease
similar to those of human brain aging, these generation. has bolstered the amyloid-β cascade hypothesis
animals generally do not readily develop the of Alzheimer’s disease28. Today, there are at least
full neuropathological or clinical phenotypes Familial cases are similar to idiopathic 25 known mutations or genomic duplications
seen in humans1,2 (with a few possible excep- cases of APP and over 170 mutations in PSEN1 or
The majority of human neurodegenera- PSEN229. Most transgenic mouse models
Mathias Jucker is at the Department of Cellular tive diseases are sporadic and of unknown overexpress mutant human APP, or PSEN1 or
Neurology, Hertie Institute for Clinical Brain etiology. However, aside from age of onset, PSEN2, or a combination of these. The overly
Research, University of Tübingen, and the DZNE- idiopathic Alzheimer’s disease, Parkinson’s hopeful expectation that the introduction
German Center for Neurodegenerative Diseases, disease, frontotemporal lobar degeneration or overexpression of wild-type or mutated
Tübingen, Germany. (FTLD) and ALS are clinically and neuro- human proteins would be sufficient to induce
e-mail: mathias.jucker@uni-tuebingen.de pathologically similar to their most common a human-like form of dementia in mice has not
familial forms15–18. Phenotypic similarities been realized.
Published online 21 September 2010; between genetic and idiopathic variants, in What these transgenic mice develop remark-
doi: 10.1038/nm.2224 combination with a paucity of natural ani- ably well, however, is cerebral β-amyloidosis,
nature medicine advance online publication 1
commentary
1999–2000
a b c d e 2004–2005
2008–2009
Number of APP-transgenic models
80
Blinded plaque load estimation
120 100 7
70 100
per study (% publications)
Number of mice per group
Gender (% publications)
100 80 60 6
(% publications)
Randomization
(% publications)
80
50 5
80 60
(median)
40 4 60
60
40 30 3
40 40
20 2
20 20
20 10 1
0 0 0 0 0
Yes NS Male Female Both NS Yes NS n=1 n≥2
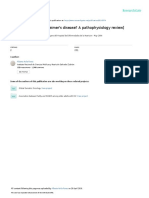
Figure 1 Studies from PubMed were retrieved using the keywords ‘mouse’, ‘amyloid’ and ‘Alzheimer’s disease’ for the years 1996–2009. Judged from the
abstracts, studies were selected that showed modulation of Aβ levels, Aβ deposition or both either through a genetic cross or through pharmacological or
nutritional interventions. Only studies in high-impact journals were selected (defined as having an impact factor >7.48), and these studies were fully and
carefully read. Shown are the data for the years 1999–2000 (n = 13), 2004–2005 (n = 43) and 2008–2009 (n = 59). (a) Less than 20% of the studies
indicate that mice have been randomly assigned to groups (NS, not specified). (b) The gender of the mice in most studies is not indicated, and mice of both
sexes were used in less than 10% of the studies. (c) The investigator seemed to be blinded toward treatment allocations and evaluation outcome in only one
third of the studies. (d) The number of mice per group varied from experiment to experiment within a study, and there were often only four to six mice per
group. (e) Two or more mouse models were examined in only 20% of the studies.
one of the hallmark pathological features of to the cognitive impairment in Alzheimer’s dis- likely that cortical synucleinopathy drives the
Alzheimer’s disease. Currently, a variety of ease remains uncertain. DLB as a complication of Parkinson’s disease
mouse models generate β-amyloid plaques, Parkinson’s disease and synucleinopathies. or as a distinct disease itself50. Thus, reports of
β-amyloid angiopathy or a mixture of both21. α-synuclein lesions are hallmarks of Parkinson’s behavioral abnormalities in α-synuclein trans-
The amyloid lesions are associated with dystro- disease, dementia with Lewy bodies (DLB) genic mice have stimulated interest in using
phic neurites, loss of dendritic spines, synaptic and multiple system atrophy. The discovery of this model to analyze nonmotor sequelae of
degeneration and robust neuroinflammation. point mutations and genomic multiplications α-synucleinopathy23.
Amyloid in the vessel wall leads to a loss of in the gene encoding α-synuclein in familial The issue of distinguishing between changes
smooth muscle cells and can lead to vascular Parkinson’s disease has sparked the genera- that cause disease and those that are comorbid is
rupture and microbleeds. All of these patholo- tion of various α-synuclein–transgenic mice24. also complicated by the recognition of familial
gies closely resemble the amyloid-associated Common haplotypes in the α-synuclein gene forms of Parkinson’s disease linked to mutations
lesions in the Alzheimer’s disease brain21,30. have now been associated with an increased in LRRK2, PARKIN, PINK1 and PARK7 (also
Despite these similarities, the commonly used risk of developing Parkinson’s disease49, known as DJ-1)18. Degeneration of nigrostri-
positron-emission tomogrophy (PET) imaging strengthening the concept of a common patho- atal dopaminergic neurons is common to all of
ligand Pittsburgh Compound B (PIB) reveals genesis of both familial and sporadic forms of these familial forms of Parkinson’s disease, but
much stronger binding to the amyloid lesions the disease. the underlying mechanisms may be distinctly
in Alzheimer’s disease brains than in mouse The overexpression of human wild-type and different, as Lewy bodies are not found in all
brains31, suggesting structural and/or biochem- mutant α-synuclein in transgenic mice leads to of these familial forms of Parkinson’s disease24.
ical differences between mouse and human synuclein lesions that resemble the Lewy bodies Mouse models exploiting these more recent
lesions32,33. PIB also binds poorly to the amy- and Lewy neurites found in Parkinson’s disease mutations have been generated, and although
loid in the nonhuman primate brain and does and DLB and the glial inclusions found in multi- these mice models have provided mechanistic
not make nonhuman primates a better model ple system atrophy22,24. α-synuclein–transgenic insights, they do not appear to show dopamin-
for this aspect of the disease34. Interestingly, mice show progressive age-dependent neuropa- ergic cell loss in the substantia nigra.
structural and biochemical β-amyloid variants thology and cognitive and locomotor dysfunc- FTLD-tau and tauopathies. The aberrant
(including poor PIB binding) have also recently tions, but the relationship of these symptoms polymerization of the microtubule-associated
been identified in the human brain35–37. to the α-synuclein lesions is not clear24. Even protein tau (MAPT) is a defining lesion in
Unlike in Alzheimer’s disease38,39, neuronal in mouse models with α-synuclein expression Alzheimer’s disease, progressive supranuclear
loss in APP-transgenic mice is modest and is confined to the substantia nigra, dopaminergic palsy, corticobasal degeneration and about
confined mainly to the hippocampus21,40,41. neuron loss in the nigrostriatal system resem- 40% of FTLD (now called FTLD-tau), includ-
Also, unlike in Alzheimer’s disease42, many bling that in Parkinson’s disease is not appar- ing Pick’s disease and frontotemporal dementia
APP-transgenic mouse lines are behaviorally ent22,50. with parkinsonism linked to chromosome 17
impaired even before they deposit Aβ in the After an initial wave of creation of (FTDP-17). Tau in the human brain is expressed
brain43,44. The findings that the infusion of α-synuclein–transgenic mice, progress in in three-repeat and four-repeat splice variants
soluble oligomeric forms of Aβ impairs cog- transgenic modeling of Parkinson’s disease in a cell type–specific manner51. In contrast,
nition45,46 and that Aβ-specific antibodies has decelerated because of uncertainty about mice express only the four-repeat tau isoforms.
and γ-secretase inhibitors rapidly attenuate what is causing the disease and what is simply Tau aggregation is dependent on the isoforms
the memory impairment of APP-transgenic related to the symptoms of Parkinson’s disease. involved, and the cell- and region-specific com-
mice47,48 support the argument that that soluble The view that Parkinson’s disease is an exclu- position of tau filaments is distinct for the vari-
Aβ species, independent of Aβ deposition, are sively motor disease affecting the dopaminergic ous tauopathies51.
sufficient to impair cognition in mice. Until the system has recently been challenged50. Whereas The discovery of MAPT mutations in FTDP-
role of soluble Aβ species in human cognitive dopaminergic neuron loss in the substantia 1752,53 initiated the generation of various trans-
decline is clarified, the relevance of behavioral nigra is the probable cause of the early-onset genic mice overexpressing mutated human tau
impairments in APP-transgenic mouse models motor problems in Parkinson’s disease, it is isoforms. These mice develop neurofibrillary
2 advance online publication nature medicine
commentary
tangles (NFTs) that resemble those seen in
FTLD-tau and Alzheimer’s disease. The initial Box 1 Predictive validity of mouse models for clinical trials
models showed high tau expression in brain- Internal validity External validity
stem and motor neurons and developed severe Randomization Models predict what they model
motor phenotypes with premature morbid- Blinded evaluation Use of several mouse models
ity. More recent models develop NFTs in the Gender and genetic background Outcome measure of clinical relevance
hippocampus and neocortex and model more Sample size and statistics Hypothesis-driven design and statistics
closely the lesions in FTLD-tau and Alzheimer’s Health status of mice Proof-of-principle versus preclinical trial
disease19, with the caveat that the NFTs in
Alzheimer’s disease and most cases of FTLD-
tau consist of wild-type tau. NFTs consisting of mice show SOD1-positive inclusions, motor induced toxicity in glial cells acts on motor
wild-type tau were successfully generated in at neuron loss and gliosis. Neurodegeneration in neurons and accelerates disease progression64.
least two studies, but these models are compli- the models is mediated through a convergence Therefore, mutant SOD1 in neurons deter-
cated by a complex genetic background54 and of several gain-of-toxic-function mechanisms mines disease onset and the early progression
the fact that NFTs appear only after 20 months acting in neurons and also through a non–cell- of disease, but the later course of the disease
of age55. Transgenic mice expressing mutated autonomous dysfunction of glial cells64. Thus, is governed instead by neighboring glial cells.
tau show cognitive impairments and neuronal the mutant SOD1 models replicate remarkably Non–cell-autonomous toxicity may also occur
death, although a discrepancy between tau- well the key clinical and neuropathological fea- in synucleinopathies and tauopathies69–72,
bearing neurons and cell death suggests that tures of this familial form of ALS. However, the and this concept has garnered more support
soluble tau or the expression of the tau trans- absence of TDP-43 inclusions in this familial by the recent findings of exogenous induc-
gene, rather than the presence of NFTs, is the variant may indicate a pathomechanism dif- tion and transcellular spread of proteopathic
neurotoxic factor25,26,56,57. ferent from idiopathic ALS, so it is possible lesions73–75.
FTLD-ALS disease spectrum and TDP-43 that treatments that work in these models may Mouse models bearing multiple transgenes
proteinopathies. TDP-43–positive cytoplasmic not be applicable to the sporadic form of the and showing multiple lesions can mimic the
inclusions are hallmarks of most sporadic ALS disease, which represents the vast majority of neuropathology of the human diseases more
and the most common subtypes of FTLD (now affected individuals. closely than do models bearing single trans-
called FTLD-TDP) and underscore the link genes (for an example, see ref. 76). However, the
between the two diseases, which is the occur- The advantages of incomplete mouse interpretation of such models is complicated by
rence of motor neuron disease in many indi- models uncertainty about the interrelationship of the
viduals with FTLD and cognitive impairment Today’s genetically engineered mouse mod- various transgenes and lesions. The observation
in many individuals with ALS27. Mutations in els recapitulate primarily the disease-defining that APP-transgenic mice do not develop NFTs
the TDP-43 gene (TARDBP) cosegregate with lesions and associated pathologies. Neuron loss and thus fail to fully model Alzheimer’s disease
both ALS and FTLD-TDP53. The majority of is apparent in some of these models, but the pathology might be seen as a disappointment.
cases of familial FTLD-TDP, however, are linked actual neurotoxic entity is often uncertain. With The mechanistic interplay between Aβ and tau
to mutations in the progranulin gene (GRN), the exception of SOD1-mutant ALS mice, ani- lesions can, however, be studied exemplarily by
underlining the heterogeneous pathomecha- mal models only partially recapitulate the com- cross-breeding APP-transgenic mice with tau-
nism of the disease27. Infrequent forms of plex clinical features of the human diseases. transgenic mice and comparing the resulting
familial ALS are caused by mutations in the Large-scale efforts are ongoing to function- offspring with the parental single-transgenic
superoxide dismutase-1 (SOD1) gene and, more ally annotate every mouse gene in the context mouse lines77–80. The enhancement of NFT
rarely, the fused in sarcoma (FUS) gene58. of the whole organism and of various environ- formation in the presence of Aβ aggregates has
The first mouse models in the new and rap- ments65, and entire pathways and organs in become a milestone observation, albeit one that
idly developing field of TDP-43 proteinopathies the mouse have already been humanized66,67. is mechanistically still unclear. Unraveling these
have shown that overexpression of both wild- Thus, the next generation of mouse models mechanisms is likely to provide prime targets
type and mutant TDP-43 can lead to TDP-43 is expected to recapitulate human neurode- for disease-modifying drugs.
inclusions as well as cortical and motor neuron generative diseases more closely. However, the
loss and therefore resemble some features of extent to which complex, human-specific neu- Incomplete mouse models in translational
the FTLD-TDP and ALS disease spectrum59,60. rodegenerative diseases can be phenotypically research
Similar to synuclein- and tau-transgenic mouse modeled in rodents remains an open question. Successful translation. An example of a rapid
models, the neurotoxic entity and the causal As it is important to distinguish disease causes and fairly predictive translation of preclinical
link between pathogenic protein and neuron from effects, incomplete models of diseases may findings into clinical studies comes from APP-
death have not been established. GRN muta- actually be advantageous models of specific dis- transgenic mice. Only four years after the gen-
tions are thought to be loss-of-function muta- ease processes. eration of the first successful APP-transgenic
tions, but the Grn-null mice analyzed thus far For example, although the spatial and tempo- mouse model81, it was reported that both
have no clear FTLD-TDP phenotype61,62. ral expression of mutated genes in the human active and passive immunization against Aβ
SOD1 mutations in ALS were first discovered brain can be modeled remarkably well using reduces cerebral amyloidosis in APP-transgenic
almost two decades ago63, and myriad transgenic genomic-based transgenic mouse models68, mice82,83. This finding has been replicated by
mutant SOD1-overexpressing mice have since mouse models using cell- and region-specific many laboratories using different mouse mod-
been generated17. These various mouse lines promoters have shown that lesions and entire els and immunization protocols84.
show progressive hindlimb tremor and weak- disease processes are non–cell autonomous. Translation of this milestone laboratory
ness, locomotor deficits and paralysis followed For instance, the cell type–specific expression finding to the clinic was remarkably rapid.
by premature death. Neuropathologically, these of mutant SOD1 has demonstrated that SOD1- In 2003, a case report provided evidence that,
nature medicine advance online publication 3
commentary
consistent with previous mouse studies, active with APP-transgenic mice (Box 1 and Fig. 1a) sion in mice, fail to slow disease progression
immunization of patients with moderate revealed that only a minority of the studies state in humans. Similarly, in light of the limited
Alzheimer’s disease reduces cerebral β-amyloid that the mice were randomly assigned to groups efficacy of intervention trials in humans with
levels, presumably through a microglia-medi- before treatment. In most studies, the gender ongoing Alzheimer’s disease, the success of
ated mechanism85. Unfortunately, the clini- of the mice was not indicated, even though Aβ prevention experiments in mice argues per-
cal trials were halted due to the unexpected deposition in mouse models is often gender suasively for preventive immunotherapy trials
occurrence of acute meningoencephalitis in dependent (Box 1 and Fig. 1b)98,99. In many in humans96.
a subset of patients86. Encephalitis was not instances, it is unclear whether the investigators
predicted by the mouse models, and only later were blinded toward treatment allocation and Need for large preclinical mouse studies
was this side effect reported to occur in mice evaluation of the outcome (Box 1 and Fig. 1c). A promising avenue to improve the predic-
under rare circumstances87,88. However, the The group sizes were often as low as four to six tive value of mouse models for clinical studies
mouse models predicted that immunization mice per group (Fig. 1d), and this number var- would be to adapt aspects of the design, quality
clears primarily parenchymal amyloid rather ied from one experiment to the next within a control and transparency of clinical studies in
than vascular amyloid and, in fact, that it may paper, raising the concern that mice were added humans.
increase cerebral amyloid angiopathy-related to a group until the desired effect was achieved For example, the internal validity of preclini-
microbleeds89,90. There is now convincing without proper statistical sample size calcula- cal mouse studies could be improved by estab-
evidence that immunization reduces cerebral tions. Rarely were two or more mouse models lishing a checklist for quality control issues
amyloid load in humans with, at least tempo- examined in any given study (Fig. 1e), and the (Box 1)97,101. In addition, a minimal standard-
rarily, an increase in cerebral amyloid angiop- health status of the mice was rarely indicated ization in methodology and quality control of
athy and microbleeds91,92. Moreover, detailed (Box 1). preclinical mouse trials for later comparisons,
postmortem analyses of immunized human The odds of ‘positive’ results are higher in interpretation and potential meta-analysis tech-
cases have found not only a reduction of amy- studies with methodological flaws97, implying niques would be desirable, including the report
loid lesions but also of the amyloid-associated that many animal studies may not withstand of negative results.
neuropathology, again showing remarkably rigorous replication. Indeed, apart from immu- The external validity of mouse studies could
similar results to those predicted from mouse nization studies of APP-transgenic mice, there benefit from including more than one mouse
studies91,93,94. Recently, vasogenic edema was are more opposing than confirmatory reports model, testing at multiple sites and using ade-
reported as an adverse side effect of passive found in the literature, although a publication quately powered designs to confirm treatment
immunization that was evident by magnetic bias toward new results cannot be excluded96,97. effects and to refute statistical flukes. Recently,
resonance imaging in humans95. The mouse A similar assessment has been made of recent for example, the extension of the lifespan of mice
counterpart of this phenomenon is currently analyses of preclinical studies using SOD1 by rapamycin was replicated simultaneously
not clear, as imaging in immunized mice has mutant mouse models of ALS100,101. at three test sites on two genetically different
rarely been done, and the neuropathological The external validity of a model refers to what mouse lines of different gender and according
features of vasogenic edema in immunized a model is good for97. However, the limitations to predetermined standardization102.
humans await further analysis. of models are rarely acknowledged, and the Most importantly, the outcome measures of
APP-transgenic mouse models of cerebral predictive validity of these models for humans preclinical studies should focus on parameters
β-amyloidosis have predicted the outcome of is often overstated or misinterpreted, raising and biomarkers with proven relevance in clini-
Alzheimer’s disease immunization trials rea- false or premature hopes for clinical efficacy. cal settings or at least those which can be tested
sonably well. Whether APP-transgenic mice APP-transgenic mice are models of cerebral in the clinic. For example, β-amyloid imaging
are useful for modeling the cognitive decline in β-amyloidosis and not of Alzheimer’s disease. and cerebrospinal fluid (CSF) measurement of
Alzheimer’s disease is less clear. If it turns out Furthermore, a treatment that is effective in Aβ and tau have become valuable biomarkers
that the current vaccination approach benefits female mice may not necessarily predict efficacy for the diagnosis and progression of Alzheimer’s
cognition in humans, the predictive value of in male subjects. Similarly, a treatment assessed disease103,104. However, few mouse studies have
the mouse models for this critical endpoint in one mouse model may not be replicable in analyzed Aβ and tau in CSF, despite the pros-
will also be affirmed (as suggested by D. Schenk another model, and the outcome measure of a pect that such studies may help to clarify the
(Elan Pharmaceutical) at the Herrenhausen treatment in a mouse model may be of no use mechanisms governing changes in protein lev-
Symposium on Neurodegeneration). for clinical translation (Box 1). els in human CSF. Rather, a primary endpoint
Failed translation? Despite the success in Therapeutic intervention studies in SOD1- of most mouse studies is postmortem neuro-
predicting important outcomes of Aβ immu- transgenic mouse models have shown that in pathology, which is arguably not very useful for
nization in Alzheimer’s disease clinical trials, it almost all instances of an apparent benefit in translational purposes.
has been argued that mouse models of human extending survival, treatment was initiated How could such large and admittedly expen-
neurodegenerative diseases have largely failed early and delayed the onset, but not the rate, sive preclinical studies be done? One option is
to predict the efficacy of clinical trials17,96. of disease progression100; nevertheless, most to establish specialized testing facilities similar
However, this apparent inconsistency appears to clinical studies have been initiated well after to the various mouse phenotype clinics that
represent less a failure of the mouse models per disease onset. Although some researchers have already exist105. Such centers could accommo-
se than a consequence of inadequate preclinical used failure of the human trials to discount the date large colonies of aged mice and use differ-
studies and misinterpretation of the models. utility of the transgenic mice, others (such as ent mouse models for validation. The inclusion
The translational failure of apparently prom- D. Cleveland (University of California–San of more aged mice in general is worth consid-
ising animal studies has at least partly been Diego) at the Herrenhausen Symposium on ering, as aging is the strongest risk factor for
attributed to the inadequate internal and exter- Neurodegeneration) argue the mouse has been neurodegenerative diseases, and neurodegen-
nal validity of preclinical studies97. Indeed, sys- a near perfect predictor of success at trial— erative disease phenotypes can differ in young
tematic analysis of the internal validity of studies treatments that fail to slow disease progres- and aged individuals50.
4 advance online publication nature medicine
commentary
Alternatively, individual laboratories could 13. Fukuhara, R., Tezuka, T. & Kageyama, T. Gene 296, & Robberecht, W. Eur. J. Neurosci. 31, 2247–2265
99–109 (2002). (2010).
seek partner laboratories and initiate inter- 14. Hamilton, B.A. Genomics 83, 739–742 (2004). 59. Wils, H. et al. Proc. Natl. Acad. Sci. USA 107, 3858–
ventions at multiple sites with different mouse 15. Ryan, N.S. & Rossor, M.N. Biomark. Med. 4, 99–112 3863 (2010).
lines and according to predefined criteria. Such (2010). 60. Wegorzewska, I., Bell, S., Cairns, N.J., Miller, T.M. &
16. Mackenzie, I.R.A. Lancet Neurol. (in the press). Baloh, R.H. Proc. Natl. Acad. Sci. USA 106, 18809–
multicenter approaches increase quality con- 17. Turner, B.J. & Talbot, K. Prog. Neurobiol. 85, 94–134 18814 (2009).
trol and have the advantage of including mice (2008). 61. Ahmed, Z. et al. Am. J. Pathol. 177, 311–324
18. Gasser, T. Expert Rev. Mol. Med. 11, e22 (2009). (2010).
housed in different environments and possibly 19. Götz, J. & Ittner, L.M. Nat. Rev. Neurosci. 9, 532–544 62. Yin, F. et al. FASEB J. published online, doi:10.1096/
mice of different health status106. In this way, (2008). fj.10-161471 (28 July 2010).
both the internal and external validity of the 20. Teschendorf, D. & Link, C.D. Mol. Neurodegener. 4, 63. Rosen, D.R. et al. Nature 362, 59–62 (1993).
38 (2009). 64. Ilieva, H., Polymenidou, M. & Cleveland, D.W. J. Cell
studies would be enhanced. However, although 21. Duyckaerts, C., Potier, M.C. & Delatour, B. Acta Biol. 187, 761–772 (2009).
the workload would be divided among labora- Neuropathol. 115, 5–38 (2008). 65. Abbott, A. Nature 465, 410 (2010).
22. Kahle, P.J. Acta Neuropathol. 115, 87–95 (2008). 66. Beckers, J., Wurst, W. & de Angelis, M.H. Nat. Rev.
tories, it may still be financially prohibitive to Genet. 10, 371–380 (2009).
23. Chesselet, M.F. Exp. Neurol. 209, 22–27 (2008).
conduct such large studies on aged animals in 24. Dawson, T.M., Ko, H.S. & Dawson, V.L. Neuron 66, 67. Traggiai, E. et al. Science 304, 104–107 (2004).
an academic university environment. 646–661 (2010). 68. Hock, B.J. Jr. & Lamb, B.T. Trends Genet. 17, S7–S12
25. Zilka, N., Korenova, M. & Novak, M. Acta Neuropathol. (2001).
Small, innovative treatment studies by indi- 118, 71–86 (2009). 69. Yazawa, I. et al. Neuron 45, 847–859 (2005).
vidual laboratories should by no means be 26. Denk, F. & Wade-Martins, R. Neurobiol. Aging 30, 70. Shults, C.W. et al. J. Neurosci. 25, 10689–10699
1–13 (2009). (2005).
replaced by large, centralized or multicenter 71. Forman, M.S. et al. J. Neurosci. 25, 3539–3550
27. Chen-Plotkin, A.S., Lee, V.M. & Trojanowski, J.Q. Nat.
preclinical mouse studies. However, the limita- Rev. Neurol. 6, 211–220 (2010). (2005).
tions of such smaller studies should be clearly 28. Hardy, J. & Selkoe, D.J. Science 297, 353–356 72. Higuchi, M. et al. J. Neurosci. 25, 9434–9443
(2002). (2005).
acknowledged by, for example, differentiating 73. Meyer-Luehmann, M. et al. Science 313, 1781–1784
29. Bertram, L. & Tanzi, R.E. Nat. Rev. Neurosci. 9, 768–
between proof-of-concept studies and preclini- 778 (2008) (2006).
74. Clavaguera, F. et al. Nat. Cell Biol. 11, 909–913
cal testing studies101. Clear editorial policies 30. Herzig, M.C., Van Nostrand, W.E. & Jucker, M. Brain
(2009).
Pathol. 16, 40–54 (2006).
established by journals on these issues, and their 31. Klunk, W.E. et al. J. Neurosci. 25, 10598–10606 75. Aguzzi, A. & Rajendran, L. Neuron 64, 783–790
enforcement by critical reviewers, will improve (2005). (2009).
76. Oddo, S. et al. Neuron 39, 409–421 (2003).
the translational value of animal studies for 32. Kuo, Y.M. et al. J. Biol. Chem. 276, 12991–12998
77. Lewis, J. et al. Science 293, 1487–1491 (2001).
(2001).
neurodegenerative diseases. 33. Maeda, J. et al. J. Neurosci. 27, 10957–10968
78. Bolmont, T. et al. Am. J. Pathol. 171, 2012–2020
(2007).
(2007).
ACKNOWLEDGMENTS 79. Terwel, D. et al. Am. J. Pathol. 172, 786–798
34. Rosen, R.F., Walker, L.C. & Levine, H. 3rd. Neurobiol.
I would like to thank L. Walker and M. Staufenbiel for (2008).
Aging published online, doi:10.1016/j.neurobio
80. Coomaraswamy, J. et al. Proc. Natl. Acad. Sci. USA
various discussions and help with this manuscript. laging.2009.02.011 (27 March 2009).
107, 7969–7974 (2010).
The systematic review of the literature by R. Radde is 35. Piccini, A. et al. J. Biol. Chem. 280, 34186–34192
81. Games, D. et al. Nature 373, 523–527 (1995).
greatly acknowledged. I also thank M. Neumann, (2005).
82. Schenk, D. et al. Nature 400, 173–177 (1999).
V. Lee, J. McLaurin, D. Schenk, D. Thal, J. Götz, 36. Levine, H. III & Walker, L.C. Neurobiol. Aging 31,
83. Bard, F. et al. Nat. Med. 6, 916–919 (2000).
542–548 (2010).
D. DiMonte, M. Goedert, T. Gasser and P. Kahle for 84. Brody, D.L. & Holtzman, D.M. Annu. Rev. Neurosci.
37. Rosen, R.F. et al. Acta Neuropathol. 119, 221–233
comments on various parts of this commentary and 31, 175–193 (2008).
(2010).
the attendees of the Herrenhausen Symposium on 85. Nicoll, J.A. et al. Nat. Med. 9, 448–452 (2003).
38. Gómez-Isla, T. et al. J. Neurosci. 16, 4491–4500 86. Orgogozo, J.M. et al. Neurology 61, 46–54 (2003).
Neurodegeneration for the inspiring discussions from (1996). 87. Furlan, R. et al. Brain 126, 285–291 (2003).
which this commentary has emerged. The work was 39. West, M.J., Coleman, P.D., Flood, D.G. & Troncoso, 88. Lee, E.B., Leng, L.Z., Lee, V.M. & Trojanowski, J.Q.
supported by the German National Genome Network J.C. Lancet 344, 769–772 (1994). FEBS Lett. 579, 2564–2568 (2005).
(NGFNPlus). 40. Calhoun, M.E. et al. Nature 395, 755–756 (1998). 89. Pfeifer, M. et al. Science 298, 1379 (2002).
41. Rupp, N.J., Wegenast-Braun, B.M., Radde, R., 90. Racke, M.M. et al. J. Neurosci. 25, 629–636
Calhoun, M.E. & Jucker, M. Neurobiol. Aging (in the (2005).
COMPETING FINANCIAL INTERESTS
press). 91. Boche, D. et al. Brain 131, 3299–3310 (2008).
The author declares no competing financial interests. 42. Morris, J.C. et al. Arch. Neurol. 66, 1469–1475 92. Rinne, J.O. et al. Lancet Neurol. 9, 363–372
(2009). (2010).
1. Walker, L.C. & Cork, L.C. in Alzheimer Disease (eds. 43. Ashe, K.H. Learn. Mem. 8, 301–308 (2001). 93. Holmes, C. et al. Lancet 372, 216–223 (2008).
R.D. Terry et al.) 233–243 (Lippincott Williams and 44. Chen, G. et al. Nature 408, 975–979 (2000). 94. Serrano-Pozo, A. et al. Brain 133, 1312–1327
Wilkins, Philadelphia, Pennsylvania, USA, 1999). 45. Lesné, S. et al. Nature 440, 352–357 (2006). (2010).
2. Gerlach, M. & Riederer, P. J. Neural Transm. 103, 46. Shankar, G.M. et al. Nat. Med. 14, 837–842 95. Salloway, S. et al. Neurology 73, 2061–2070
987–1041 (1996). (2008). (2009).
3. Awano, T. et al. Proc. Natl. Acad. Sci. USA 106, 47. Dodart, J.C. et al. Nat. Neurosci. 5, 452–457 96. Zahs, K.R. & Ashe, K.H. Trends Neurosci. 33, 381–
2794–2799 (2009). (2002). 389 (2010).
4. Cork, L.C. et al. J. Neuropathol. Exp. Neurol. 47, 48. Comery, T.A. et al. J. Neurosci. 25, 8898–8902 97. van der Worp, H.B. et al. PLoS Med. 7, e1000245
629–641 (1988). (2005). (2010).
5. Price, D.L. et al. Brain Pathol. 1, 287–296 (1991). 49. Simón-Sánchez, J. et al. Nat. Genet. 41, 1308–1312 98. Callahan, M.J. et al. Am. J. Pathol. 158, 1173–1177
6. Cummings, B.J., Su, J.H., Cotman, C.W., White, (2009). (2001).
R. & Russell, M. J. Neurobiol. Aging 14, 547–560 50. Obeso, J.A. et al. Nat. Med. 16, 653–661 (2010). 99. Wang, J., Tanila, H., Puolivali, J., Kadish, I. & van
(1993). 51. Goedert, M. & Spillantini, M.G. Science 314, 777–781 Groen, T. Neurobiol. Dis. 14, 318–327 (2003).
7. Walker, L.C. Brain Res. Brain Res. Rev. 25, 70–84 (2006). 100. Benatar, M. Neurobiol. Dis. 26, 1–13 (2007).
(1997). 52. Heutink, P. Hum. Mol. Genet. 9, 979–986 (2000). 101. Ludolph, A.C. et al. Amyotroph. Lateral Scler. 11,
8. Nelson, P.T., Greenberg, S.G. & Saper, C.B. Neurosci. 53. Sleegers, K., Cruts, M. & Van Broeckhoven, C. Annu. 38–45 (2010).
Lett. 170, 187–190 (1994). Rev. Neurosci. 33, 71–88 (2010). 102. Harrison, D.E. et al. Nature 460, 392–395 (2009).
9. Schultz, C., Hubbard, G.B., Tredici, K.D., Braak, E. & 54. Andorfer, C. et al. J. Neurochem. 86, 582–590 103. Blennow, K. Nat. Med. published online, doi:10.1038/
Braak, H. Adv. Exp. Med. Biol. 487, 59–69 (2001). (2003). nm.2221 (21 September 2010).
10. Rosen, R.F. et al. J. Comp. Neurol. 509, 259–270 55. Ishihara, T. et al. Am. J. Pathol. 158, 555–562 104. Perrin, R.J., Fagan, A.M. & Holtzman, D.M. Nature
(2008). (2001). 461, 916–922 (2009).
11. Holzer, M., Craxton, M., Jakes, R., Arendt, T. & 56. Santacruz, K. et al. Science 309, 476–481 (2005). 105. Fuchs, H. et al. Curr. Pharm. Biotechnol. 10, 236–243
Goedert, M. Gene 341, 313–322 (2004). 57. de Calignon, A. et al. Nature 464, 1201–1204 (2009).
12. Podlisny, M.B., Tolan, D.R. & Selkoe, D.J. Am. J. (2010). 106. Martin, B., Ji, S., Maudsley, S. & Mattson, M.P. Proc.
Pathol. 138, 1423–1435 (1991). 58. Bento-Abreu, A., Van Damme, P., Van Den Bosch, L. Natl. Acad. Sci. USA 107, 6127–6133 (2010).
nature medicine advance online publication 5
C O M M E N TA RY
The future of genetic research on
neurodegeneration
Christine Van Broeckhoven
Why, with all the progress in the field Yet, at least in the case of Alzheimer’s disease, receptor 1 (CR1) and phosphatidylinositol-
of neurodegeneration, do we still lack these early days of so-called ‘low-hanging fruit’ binding clathrin assembly protein (PICALM))
disease-modifying drugs that tackle the were followed by nearly 15 years without real previously undescribed risk genes12,13. Another
primary defect of severe cell loss? How breakthroughs in genetics. Linkage analyses in approach is to homogenize the study popula-
much progress has been made toward individuals with familial late-onset Alzheimer’s tion on the basis of a shared characteristic. This
this goal? Have we spent our time and disease revealed chromosomal regions (on chro- was done in a GWAS on FTLD characterized
resources wisely? And, most important, mosomes 9, 10 and 12) that probably harbor a by pathology involving the TAR DNA-binding
is there room for improvement? This gene associated with Alzheimer’s disease9, but, protein TDP43, and led to the identification
commentary highlights several problems ten years later, the genes explaining the observed of what might be the first risk gene for FTLD,
faced by researchers in studying the linkage signals have yet to be found. A flurry transmembrane protein-106B (TMEM106B)14.
genetic etiology of neurodegenerative of candidate gene–based association studies To obtain a sufficient number of subjects for
diseases and seeks to provide direction were published, but for every positive report, statistical analyses, several concessions were
in overcoming some of these obstacles. negative replication studies followed. Even the made in this study14. Clustering subjects by
initial wave of high-density chromosome- and pathology meant that the study population
Over the past two decades the field of molecu- genome-wide association studies (GWASs) on was more variable than ideal in terms of clini-
lar genetics has contributed substantially to Alzheimer’s disease failed to deliver substan- cal presentation, population background and
our understanding of neurodegeneration. tial insight into the disease. Slight differences inclusion of carriers of highly penetrant muta-
The genetic basis of neurodegenerative dis- in design notwithstanding, the only convinc- tions in genes giving rise to TDP43 pathology.
eases such as Huntington’s disease, Alzheimer’s ing association to be detected in these studies It is promising, though, that studies including
disease, Parkinson’s disease, frontotemporal was with apolipoprotein E (APOE) variant ε4, such a heterogeneous group of subjects and
lobar degeneration (FTLD) and amyotrophic the single widely accepted genetic risk factor controls have yielded putative risk genes.
lateral sclerosis (ALS) is no longer questioned. for Alzheimer’s disease. Although an optimist If we continue down this road, what will
Genetic analysis of multiple generations of could interpret this as a proof of concept, it is come next? A further increase in sample size
affected families paved the way in identifying distressing that all these efforts and resources allows the detection of polymorphisms with
new pathways and pathogenic cascades. For only produced confirmatory evidence for a risk even smaller effects, as foreshadowed by meta-
instance, in Alzheimer’s disease, a consider- factor that was identified in 1993 (ref. 10). analyses in other complex traits15. Analysis of
able proportion of our current understanding epistasis and pathophysiological pathways has
of the pathogenesis originates from genetic New approaches for genetic analyses the potential to uncover additional small genetic
breakthroughs in the early 1990s in a handful Many obstacles have contributed to this slow effects. But will these advances translate into
of families with a rare early-onset dominant progress. A central concern is the inherent improved patient care? The first genome-wide
form of the disease1–8. difference between identifying a pathogenic single nucleotide polymorphism (SNP)-based
mutation that segregates with disease in a pathway analyses on Alzheimer’s disease16,17
monogenic family and finding a genetic risk (presented at the International Conference on
Christine Van Broeckhoven is at the factor that explains no more than 1% of the Alzheimer’s Disease 2010) did not reveal new
Neurodegenerative Brain Diseases Group, heritability of a trait in a population11. An pathomechanisms but rather reiterated long-
Department of Molecular Genetics, VIB, Antwerpen, intuitive approach (albeit difficult to imple- standing hypotheses about the involvement of
Belgium, and the Laboratory of Neurogenetics, ment) in identifying risk factors with small lipid metabolism and the immune response
Institute Born-Bunge, University of Antwerp, effect sizes is to increase the size of the study in the pathogenesis of Alzheimer’s disease.
Antwerpen, Belgium. population. In Alzheimer’s disease, thousands Nevertheless, earlier attempts to target these
e-mail: christine.vanbroeckhoven@molgen.vib-ua.be of samples from different populations and pathways pharmacologically proved unsuc-
study designs have been pulled together, which cessful18,19. Arguably, genetic profiling on the
Published online 21 September 2010; led to the identification of at least one (clus- basis of underlying patterns of susceptibility
doi: 10.1038/nm.2225 terin (CLU)), but probably three (complement might help to streamline the design of future
nature medicine advance online publication 1
commentary
clinical trials by including subjects who would According to the Catalog of Published Genome- repeatedly detected in individuals with a clinical
benefit most from the compound under study, Wide Association Studies (http://www.genome. phenotype of Alzheimer’s disease24, a preseni-
but in reality we have not advanced sufficiently gov/gwastudies/), more than 50 genome-wide lin-1 (PS1) mutation has been detected in an
to implement these tools in such a way. significant hits for a wide range of traits have individual with autopsy-confirmed Pick’s dis-
been found at intergenic SNPs, substantiating ease25 and, for mutations in the gene encoding
The complexity of genetic risk the notion that these regions may harbor as yet progranulin (GRN), clinical heterogeneity is
Geneticists often argue that finding a genetic unknown functional elements. Experimental frequently reported26. It is not inconceivable
risk factor of small effect size does not neces- confirmation of small genetic effects is chal- that intermediate to low penetrant susceptibil-
sarily have to translate directly into an advance lenging enough in and of itself. It becomes even ity alleles also contribute to more than one of
in patient care. Rather, it may bring to light more daunting in the absence of any obvious the clinically distinct syndromes, perhaps by
important pathways for further study through testable hypothesis. Experimental modeling of accelerating the neurodegenerative cascade
cell biology and to incorporate into new animal epistatic networks and long-range effects adds downstream of aberrant protein aggregation.
models of disease. another layer of complexity. At this rate, transla- This raises the question of whether we should
There are several issues with this notion. First, tion into meaningful biomarkers or therapeutic perform our studies across the clinical diag-
at what point are we sufficiently convinced that approaches will still be a long way off. nostic boundaries. The need to define an indi-
a newly described genetic risk factor or pathway The absence of a clear biological interpre- vidual’s phenotype is different from a clinical
warrants further investigation? Most research- tation does not diminish the relevance of a perspective (at least in the absence of therapeu-
ers are reluctant ‘believers’ in genes that confer finding. There are several opportunities for tics targeting the underlying causes) than from
a relatively small increase in risk observed in geneticists to validate their findings and provide a research perspective. By studying endopheno-
association studies, in part because the high more depth to their observations. Apart from types (for instance, TDP-43 protein abundance,
odds ratios obtained in genetic association an obvious need for better annotation of the cortical thickness or age of onset) regardless of
studies of APOE ε4 unrealistically raised our genome in a multitude of cell types (an ongoing the clinical diagnosis, disease modifiers may
expectations, and because too many attempts effort at the RIKEN Institute in Japan), genetic be discovered that can ultimately provide suit-
to find additional risk genes have failed. This data can be combined with data generated from able targets for therapeutic intervention for a
is true even when the association is observed proteomics or transcriptomics experiments in broader range of patients.
consistently across studies and populations. For samples from the same individuals. Additional
instance, genetic variability in sortilin-related data can also be collected on other markers of Reexamining early-onset forms of disease
receptor, L(DLR class) A repeats-containing disease progression, including cerebrospinal Undescribed genetic loci could also be identi-
(SORL1) has been repeatedly associated with fluid (CSF) biomarkers, structural measures fied by refocusing attention and resources on
Alzheimer’s disease in populations of European, such as hippocampal atrophy or ventricular unexplained early-onset forms of Alzheimer’s
African, Latino and Asian ancestry (http://www. enlargement and measures of cognition, to disease. Perhaps we are not putting all the avail-
alzgene.org/). This strongly suggests that SORL1 identify underlying pathways or pathomecha- able resources for gene finding to use. Although
is involved in the disease. Nevertheless, SORL1 is nisms and to increase confidence in the associa- genetic research on Alzheimer’s disease in the
not widely acknowledged as a genuine risk gene tion of genetic variants in these risk genes with past decade has largely concentrated on the
for Alzheimer’s disease. As a consequence of this disease21–23. This calls for longitudinal collec- complexity of late-onset disease and its low-
hesitation to trust new associations, few genes tion of broad phenotypic data (including envi- penetrant risk factors, much of the familial
are carried forward into in-depth molecular ronmental exposure, early-life history of disease clustering in early-onset Alzheimer’s disease
genetic analyses aimed at identifying function- and medication), as well as repeated systematic is still enigmatic. Prevalence estimates sug-
ally relevant sequence variants that can serve as biosampling, not only of DNA and RNA, but gest that more than 10% of individuals with
starting points for in vivo studies. also of body fluids, skin (for induced pluri- Alzheimer’s disease have early onset disease, but
Second, the pathways that are brought to potent stem cell studies) and brain at autopsy. only a limited proportion can be explained by
light, although interesting, are difficult to This might require studies to be scaled down in known mutations27, indicating that additional
study through reductionist molecular biol- terms of sample size, but if the design includes studies of early-onset Alzheimer’s disease using
ogy techniques. For example, take clusterin, a enrichment for genetic factors (for instance, an current high-throughput genetic tools might
chaperone molecule that is involved in a wide early-onset cohort or a genetically isolated pop- prove fruitful. By analogy, recent progress in the
range of physiological processes, any number of ulation), and a willingness to collaborate among genetics of FTLD and ALS has been driven by
which could be disturbed in Alzheimer’s disease. the researchers, the chance of identifying robust family-based studies28–34. These families might
Moreover, these processes are not always linear; genetic associations increases. The availability of also provide a suitable setting in which to dis-
depending on the ratio of clusterin to amyloid-β biosamples will also enable a more rapid trans- cover genetic modifying factors, which perhaps
for instance, clusterin can have either proamy- lation of findings back to the subject. delay onset or explain intrafamilial heterogene-
loidogenic or neuroprotective effects20. For now, A related point of consideration in the design ity in clinical presentation.
it remains undecided whether physiological of genetic studies derives from the growing With technological advances in whole-
effects of risk alleles can be observed in a sim- awareness that the various neurodegenerative exome and genome sequencing, multiplex
ple cellular model or whether they require more diseases show marked overlap at the patho- families in which the disease-related mutation
complex modeling in a more natural context. logical level, which suggests that they share is still unidentified because of broad linkage
Third, the situation is complicated fur- pathological mechanisms. Pathogenic muta- peaks or the absence of simple coding muta-
ther when (as is often the case) the strongest tions in genes that are generally considered to tions in regions of interest35 can be resolved by
association is observed for a SNP without cause one type of neurodegenerative disease sequencing three or four affected relatives. This
an obvious effect on the protein function or have been identified in people clinically diag- approach might also be applied to less infor-
expression, in genes of unknown function nosed with another type of neurodegeneration. mative families (in which most relatives are
(such as TMEM106B) or in intergenic regions. A mutation in the FTLD gene tau (MAPT) is no longer available for linkage analysis) or to
2 advance online publication nature medicine
commentary
identify the presence of one or more variants gene encoding amyloid precursor protein (APP) contributors, however, are the many people with
of intermediate penetrance. When applied to in people with Alzheimer’s disease41,42 support neurodegenerative disease and their families that
generously participate in research and the efforts of
risk genes identified from GWASs, clustering this contention and suggest that rare, recessive researchers to pave the way toward new therapeutics.
of trait-associated rare and common variants mutations in other genes may contribute to
in functional domains in these genes could ‘sporadic’ Alzheimer’s disease. These mutations COMPETING FINANCIAL INTERESTS
suggest new disease mechanisms. By analogy, can be traced in GWAS genotype data43,44 or by The author declares no competing financial interests
the observation of a predominant location of whole-genome sequencing of selected individu- 1. St. George-Hyslop, P. et al. Nat. Genet. 2, 330–334
pathogenic mutations in the C-terminal regions als with the disease, possibly in genetically iso- (1992).
2. St. George-Hyslop, P. et al. Science 235, 885–890
of TARDBP and FUS suggests a role of aberrant lated populations. In addition, other sources of (1987).
RNA processing in neurodegeneration36. genetic variation remain to be explored. Copy 3. Goate, A. et al. Nature 349, 704–706 (1991).
The possibility that rare variants of intermedi- number variation and mutations in regula- 4. Sherrington, R. et al. Nature 375, 754–760 (1995).
5. Van Broeckhoven, C. et al. Nat. Genet. 2, 335–339
ate penetrance might be involved in Alzheimer’s tory regions are more readily investigated in (1992).
disease has received limited attention but war- Parkinson’s disease45, but there is evidence 6. Levy-Lahad, E. et al. Science 269, 973–977 (1995).
rants further investigation. For example, null that gene dosage effects are involved in other 7. Rogaev, E.I. et al. Nature 376, 775–778 (1995).
8. Van Broeckhoven, C. et al. Science 248, 1120–1122
mutations in GRN are a highly penetrant cause neurodegenerative conditions as well, includ- (1990).
of FTLD28,29, but GRN missense mutations are ing APP duplications in Alzheimer’s disease46 9. Bertram, L. & Tanzi, R.E. Hum. Mol. Genet. 13 Spec No
also observed at an increased frequency in indi- and MAPT duplication47 and GRN deletions 1, R135–R141 (2004).
10. Corder, E.H. et al. Science 261, 921–923 (1993).
viduals with FTLD and Alzheimer’s disease37,38. in FTLD48, as well as promoter mutations and 11. Sleegers, K. et al. Trends Genet. 26, 84–93 (2010).
The precise relevance of these GRN missense mutations that affect miRNAs or their binding 12. Harold, D. et al. Nat. Genet. 41, 1088–1093 (2009).
13. Lambert, J.C. et al. Nat. Genet. 41, 1094–1099
mutations to neurodegeneration is still an area sites49. The first genome-wide studies on copy- (2009).
of active investigation, but in silico and in vitro number variation in Alzheimer’s disease are 14. Van Deerlin, V.M. et al. Nat. Genet. 42, 234–239
evidence suggests that these mutations may ongoing, and they may reveal more common (2010).
15. Teslovich, T.M. et al. Nature 466, 707–713 (2010).
impair progranulin function and affect suscep- copy number polymorphisms that affect risk 16. Lambert, J.C. et al. J. Alzheimers Dis. 20, 1107–1118
tibility to neurodegeneration26. Interestingly, (for example, ref. 50). (2010).
serum progranulin concentrations in some Additional questions remain. What are the 17. Jones, L. et al. Alzheimers Dement. 6, S113 (2010).
18. Aisen, P.S. Lancet Neurol. 1, 279–284 (2002).
people carrying a GRN missense mutation are implications of alternative splicing, distant 19. Trompet, S. et al. J. Neurol. 257, 85–90 (2010).
intermediate between null mutation carriers regulatory elements or antisense transcription 20. Yerbury, J.J. et al. FASEB J. 21, 2312–2322 (2007).
21. Kauwe, J.S. et al. J. Alzheimers Dis. published online,
and nonmutated control samples39, underscor- for disease susceptibility? How important is doi:10.3233/JAD-2010-091711 (15 July 2010).
ing the idea that these variants might act as risk epigenetics in neurodegeneration? What is the 22. Potkin, S.G. et al. PLoS One 4, e6501 (2009).
factors of intermediate effect. This emphasizes contribution of somatic mutations to the occur- 23. Shulman, J.M. et al. PLoS One 5, e11244 (2010).
24. Rademakers, R. et al. Hum. Mutat. 22, 409–411
the pathogenic relevance of rare sequence rence of disease? Is it sufficient to sequence DNA (2003).
variants that do not fit in the picture of what is extracted from lymphocytes? What could we 25. Dermaut, B. et al. Ann. Neurol. 55, 617–626 (2004).
considered pathogenic (in the case of GRN-null learn from sequence analysis or RNA profiling 26. Sleegers, K., Cruts, M. & Van Broeckhoven, C. Annu.
Rev. Neurosci. 33, 71–88 (2010).
mutations). in the aging brain? 27. Bettens, K., Sleegers, K. & Van Broeckhoven, C. Hum.
The first wave of next-generation sequencing These are just some of the questions we can Mol. Genet. 19, R4–R11 (2010).
28. Baker, M. et al. Nature 442, 916–919 (2006).
efforts is ongoing40. It is imperative to carefully ask ourselves, and for many we have no answer 29. Cruts, M. et al. Nature 442, 920–924 (2006).
design these studies and not to move forward yet. But whether setting out to explore these 30. Van Deerlin, V.M. et al. Lancet Neurol. 7, 409–416
simply because the technique and the money are less well-defined concepts, or following more (2008).
31. Sreedharan, J. et al. Science 319, 1668–1672
available. Such studies require careful thought conventional approaches, we should try to stay (2008).
on which subjects to select, which source of close enough to the affected individual and 32. Kabashi, E. et al. Nat. Genet. 40, 572–574 (2008).
DNA to use and which technique to employ his or her disease, by incorporating strategies 33. Kwiatkowski, T.J. Jr. et al. Science 323, 1205–1208
(2009).
(that is, do we expect only amino acid substi- for phenotyping and biobanking in the study 34. Vance, C. et al. Science 323, 1208–1211 (2009).
tutions or nonsense mutations?). And given the design in close collaboration with clinicians. 35. Rademakers, R. et al. Am. J. Hum. Genet. 77, 643–652
(2005).
many sequence variations that will be detected, This allows a rapid initial assessment of the 36. Sleegers, K. & Van Broeckhoven, C. Nature 458, 415–
how do we go about separating the wheat from biological relevance of the genetic findings in 417 (2009).
the chaff? The challenges that follow sequenc- biosamples, as well as an exploration of gen- 37. Brouwers, N. et al. Neurology 71, 656–664 (2008).
38. van der Zee, J. et al. Hum. Mutat. 28, 416 (2007).
ing should not be underestimated. We should otype-phenotype correlations, putative bio- 39. Sleegers, K. et al. Ann. Neurol. 65, 603–609 (2009).
be prepared for the possibility that these tech- markers or the effect of compounds on human 40. Zuchner, S., Beecham, G., Haines, J. & Pericak-Vance,
niques uncover a different variety of genetic risk material, in a first attempt to translate the find- M.A. Alzheimers Dement. 6, S74 (2010).
41. Di Fede, G. et al. Science 323, 1473–1477 (2009).
variants that cannot easily be modeled with the ings to patient care. 42. Tomiyama, T. et al. Ann. Neurol. 63, 377–387 (2008).
current approaches. Proving causality could be a 43. Clarimón, J. et al. Neurobiol. Aging 30, 1986–1991
ACKNOWLEDGMENTS (2009).
bottleneck. The availability of extended (endo-) A number of the issues identified in this commentary 44. Nalls, M.A. et al. Neurogenetics 10, 183–190 (2009).
phenotypic data and biosamples may facilitate were discussed in a session on genetics at the 45. Nuytemans, K., Theuns, J., Cruts, M. & Van Broeckhoven,
the process of establishing the true pathogenic Herrenhausen Symposium on Neurodegeneration C. Hum. Mutat. 31, 763–780 (2010).
with P. St. George-Hyslop, P. Heutink and 46. Rovelet-Lecrux, A. et al. Nat. Genet. 38, 24–26
variants, by allowing genotype-phenotype cor- B. De Strooper, with added discussion points and (2006).
relation and functional characterization of the commentary provided by the attendees at the 47. Rovelet-Lecrux, A. et al. J. Alzheimers Dis. published
detected variants directly in biosamples from conference. The research in my group has been online, doi:10.3233/JAD-2010-100441 (15 July
2010).
affected individuals. funded by the Flemish and Federal governments of
48. Gijselinck, I. et al. Hum. Mutat. 29, 53–58 (2008).
Belgium, the VIB, the Institute Born-Bunge and the 49. Theuns, J. et al. Am. J. Hum. Genet. 78, 936–946
The possibility of recessive mutations in University of Antwerp, as well as by many national (2006).
Alzheimer’s disease is basically unexplored. Two and international governmental research funding 50. Beecham, G.W. et al. Alzheimers Dement. 6, S113
recent studies on homozygous mutations in the agencies and charity organizations. The main (2010).
nature medicine advance online publication 3
C O M M E N TA RY
Biomarkers in Alzheimer’s disease drug
development
Kaj Blennow
Biomarkers may be of great value in clinical trials and may have disease-modifying Alzheimer’s disease cases with advanced neu-
Alzheimer’s disease drug development to effects. The vast majority of these new treat- rodegeneration. A potential example of this
select the most optimal drug candidates ments aim to inhibit Aβ toxicity and include is the follow-up study of cases in the Elan
for large and expensive phase 3 clinical secretase inhibitors inhibiting the production AN1792 phase 1 Aβ immunotherapy trial. In
trials. Biomarkers will also be important of Aβ from APP, immunotherapies aimed at subjects who underwent post-mortem assess-
to provide evidence that a drug affects increasing the clearance of Aβ from the brain ment, there was evidence of marked plaque
the underlying pathophysiology of the and inhibitors of Aβ aggregation1. If this type removal, despite their clinical deterioration to
disease, which, together with a beneficial of anti-Aβ treatment proves able to reduce severe dementia before death4. Thus, arrest-
effect on the clinical course, will be plaque pathology and have a beneficial effect ing Aβ aggregation or even plaque removal
essential for labeling the drug as having on cognition in people with Alzheimer’s dis- may have limited effect in Alzheimer’s disease
a disease-modifying effect. ease, then the recent years of Alzheimer’s dis- cases with dementia, owing to severe neuronal
ease research will be a success story. and synaptic loss and heavy tangle load at this
Twenty-five years ago, our knowledge of the Unfortunately, reports of clinical trials stage of disease.
molecular pathogenesis of Alzheimer’s disease showing negative results have begun to accu- It is likely that we need to design trials on the
was very limited. In the mid-1980s, researchers mulate. In recent years, there have been sev- basis of diagnostic algorithms that recognize
succeeded in identifying amyloid-β (Aβ) and eral clinical trials where drug candidates with the predementia stage (prodromal Alzheimer’s
tau as the main components of plaques and an Aβ-targeting mode of action have failed to disease) or even the asymptomatic phase of
tangles, which are the hallmarks of the dis- show any substantial effects on primary cog- the disease (preclinical Alzheimer’s disease)
ease. Together, these important achievements nitive outcome measures, such as tramipro- to give promising drug candidates a chance
marked the start of modern Alzheimer’s dis- sate (that binds soluble Aβ, thus maintaining of showing a disease-modifying effect. This
ease research, and subsequent research efforts it in a nonfibrillar form) and tarenflurbil (a proposal is supported by studies in transgenic
have led to a detailed knowledge of amyloid γ-secretase modulator)3. These disappoint- mouse models of Alzheimer’s disease, which
precursor protein (APP) metabolism, Aβ gen- ing results are causing concern that the amy- suggest that drugs would be most efficacious
eration and the pathogenic events involved in loid cascade hypothesis will be proved wrong early on in the disease process, before severe
plaque development and of tau homeostasis and that the potential success will turn into plaque pathology is present5.
and tangle formation1. The prevailing hypoth- a failure. However, there are several other Mild cognitive impairment (MCI) may be
esis for Alzheimer’s disease pathogenesis is the possible explanations for this. These include regarded as a transitional state between nor-
amyloid cascade hypothesis, which states that that the trials have been done in subjects with mal aging and Alzheimer’s disease. However,
Aβ aggregation with plaque formation is the Alzheimer’s disease with dementia (that is, at MCI is an etiologically heterogeneous entity;
central event, ultimately leading to neuronal a far too advanced stage of the disease), that only ~50% of people with MCI have pro-
degeneration and clinical symptoms2. clinical diagnostic procedures have been too dromal Alzheimer’s disease, whereas others
These research advances have led to a large unspecific to allow identification of a homo have a benign form of MCI as part of the
number of drug candidates that are now in genous group of patients with Alzheimer’s dis- normal aging process and some have other
ease pathology and that the drug candidates disorders6. Given that individuals with MCI
Kaj Blennow is at the Clinical Neurochemistry tested have been ineffective despite promising, have only mild disturbances in episodic
Laboratory, Institute of Neuroscience and but misleading, data from preclinical develop- memory and other characteristic symptoms
Physiology, Department of Psychiatry and ment. of Alzheimer’s disease are absent or vague, it
Neurochemistry, The Sahlgrenska Academy at is a great challenge for the clinician to identify
University of Gothenburg, Mölndal, Sweden. Are we too far down the road in clinical which MCI cases have prodromal Alzheimer’s
e-mail: kaj.blennow@neuro.gu.se trials? disease.
It is logical to assume that disease-modify- Testing Alzheimer’s disease drugs in clini-
Published online 21 September 2010; ing drugs targeting Aβ will have only minor cal trials in which unspecified MCI cases are
doi: 10.1038/nm.2221 effects on the clinical course of the disease in included will thus mean that around half of
nature medicine advance online publication 1
commentary
Position as theragnostic
the cases do not have prodromal Alzheimer’s biomarker in trial type
disease, the disorder for which the drug is Aβ
intended, which may seriously affect the pos- Aβ immuno- BACE1 γ-secretase aggregation
Biomarker Pathogenic process therapy inhibitor inhibitor inhibitor
sibility of identifying clinical effects. One pos-
sible example so far is the large trial on the Amyloid PET Brain Aβ load
cholinesterase inhibitor donepezil, in which
more than 700 subjects with unspecified MCI CSF Aβ42
γ-secretase-dependent
were treated for 3 years without any signifi- APP and Aβ metabolism
CSF Aβ40
Primary biomarkers
cant effect of donepezil treatment7.
The addition of positive biomarkers as an
inclusion criterion in such MCI trials would CSF sAPPβ
β-secretase-dependent
enrich the proportion of subjects with under- CSF BACE APP and Aβ metabolism
lying Alzheimer’s disease pathology and, activity
thereby, increase the possibility of identifying CSF Aβ1–14,
a positive effect of a drug. Several studies have Aβ1–15, Aβ1–16 γ-secretase-independent
APP and Aβ metabolism
consistently shown that the combination of CSF sAPPα
high cerebrospinal fluid (CSF) levels of total
tau (T-tau) and phosphorylated tau (P-tau) CSF Aβ oligomers Aβ oligomerization
and low levels of the 42–amino-acid isoform
of Aβ (Aβ42) has a high predictive value for CSF total tau Intensity of neuronal
degeneration and
identifying cases of prodromal Alzheimer’s
Downstream
biomarkers
MRI hippocampal brain atrophy rate
disease in people with MCI, with one study volume
reporting a sensitivity of 95%8. A high pre- Tau phosphorylation
CSF phospho-tau
dictive value has been verified in large mul- and tangles
ticenter studies, including the Alzheimer’s Brain glucose
FDG PET metabolism
Disease Neuroimaging Initiative study9, the
Descripa study10 and the Swedish Brain Power
Figure 1 Flow chart for hypothetical use of theragnostic biomarkers in Aβ-targeting clinical trials.
project11. Numerous studies have also shown
Theragnostic biomarkers can be divided into primary biomarkers (orange), used to identify and monitor
that magnetic resonance imaging (MRI) the specific biochemical effect, or mode of action, of the drug, and downstream biomarkers (blue),
measurement of hippocampal atrophy and used to identify and monitor effects on pathogenic processes downstream of the drug target. At right,
positron emission tomography (PET) imag- validated biomarkers that may be valuable to monitor specific pathogenic processes are indicated with
ing with the 2-[18F]fluoro-2-deoxy-d-glucose green boxes, whereas promising biomarkers that need further development and validation of methods
(FDG) ligand and β-amyloid–binding trac- are indicated with yellow boxes. sAPP, soluble APP.
ers such as Pittsburgh Compound-B (PiB)
have high predictive values of approximately degree of noise into trials, which may mark- (the number or extent of Aβ plaques in the
85–90% for prodromal Alzheimer’s disease in edly reduce the possibility of identifying any brains of Alzheimer’s disease–transgenic
the MCI stage of the disease12,13. Thus, bio- effect of a drug. mice) but have no beneficial effect in people
markers will be key tools for enrichment of It is likely that the effectiveness of disease- with Alzheimer’s disease, indicating that these
clinical trials with true prodromal Alzheimer’s modifying drugs targeting Aβ will vary mouse models have a very low predictive
disease cases. between cases depending on the severity of power of treatment success in patients with
the Aβ plaque pathology. In other words, sporadic Alzheimer’s disease1. Alzheimer’s
Is Alzheimer’s disease too clinically diagnosed Alzheimer’s disease cases disease–transgenic mice have a huge over-
heterogeneous? with biomarker evidence of a disturbance in expression of Aβ and develop plaques much
Trials involving clinically diagnosed sub- Aβ metabolism—for example, low CSF Aβ42 faster (months instead of decades) than
jects with Alzheimer’s disease may enroll or high PiB binding on PET—may be more people who develop Alzheimer’s disease and
patients with different pathologies, owing responsive to anti-Aβ drugs than individuals are thus probably much more responsive to
to misdiagnoses. Apart from the fact that not showing such a disturbance. Biomarkers anti-Aβ treatment than humans with sporadic
the relative intensity of plaques and tangles reflecting the central pathogenic processes Alzheimer’s disease. Indeed, several anti-Aβ
may differ markedly among individuals with in Alzheimer’s disease may thus be valuable drug candidates that were shown to reduce
Alzheimer’s disease14, there is also a sizable for post hoc data analyses of clinical trials to Aβ burden in Alzheimer’s disease–transgenic
overlap in pathology between Alzheimer’s stratify patient cohorts, enabling identifica- mice, such as tramiprosate (an Aβ antiaggre-
disease and other dementias, such as Lewy tion of subgroups with more homogeneous gation compound), tarenflurbil (a γ-secretase
body dementia and vascular dementia15,16. neuropathology. modulator), rosiglitazone (a type 2 diabetes
Current clinical criteria for Alzheimer’s dis- drug that also acts as a β-secretase inhibitor),
ease may also be too nonspecific to identify Are we taking poor drug candidates into statins, nicotine, phenserine (a cholinest-
cases of pure Alzheimer’s disease with high phase 3 clinical trials? erase inhibitor that also reduces Aβ amounts
certainty. For example, recent data show that The effect of disease-modifying, Aβ-targeting by decreasing APP mRNA translation) and
approximately 20% of clinically diagnosed drug candidates on plaque pathology is com- estrogens, have now failed in clinical trials of
Alzheimer’s disease cases enrolled in clini- monly evaluated in APP and presenilin trans- patients with Alzheimer’s disease1,3. It is likely
cal trials may have negative PiB-PET scans17. genic mice. There are numerous drugs that that the promising data from the Alzheimer’s
This diagnostic uncertainty introduces a high cause a substantial reduction in ‘Aβ burden’ disease–transgenic mice models may have had
2 advance online publication nature medicine
commentary
a large impact on decisions to proceed with tion in a manner that correlates well with production. Further information on the qual-
these drugs to phase 3 clinical trials. both neuropathological measures of tangle ity control program is available at http://www.
One example of this might be tarenflurbil, load and cognitive symptoms12. Last, PET neurochem.gu.se/TheAlzAssQCprogram/.
which is the R-enantiomer derivative of flur- imaging with the FDG ligand allows for the For structural MRI imaging of medial tem-
biprofen, a nonsteroidal anti-inflammatory assessment of the glucose metabolism rate poral lobe atrophy, there is no consensus on
drug (NSAID). Data from experiments in cell in specific brain regions, whereas fibrillar Aβ which structure is the region of interest (hip-
culture and in Alzheimer’s disease–transgenic deposits and plaques in the brain can be visu- pocampus, amygdala, entorhinal cortex or
mice suggest that this drug selectively lowers alized with β-amyloid–binding tracers such as parahippocampal gyrus), whether to use visual
Aβ42 amounts and acts as a γ-secretase mod- PiB13. These biomarkers may serve as valuable rating or computerized algorithms for evalu-
ulator18. The authors of this study cautiously theragnostic tools in Alzheimer’s disease drug ation of atrophy and whether age-dependent
concluded that “rigorous clinical testing of development. cutoffs should be applied for hippocampal
R-flurbiprofen will be necessary to determine Treatment with symptomatic drugs (such atrophy to correct for age-related changes12.
if it has the ability to lower Aβ42 in humans as cholinesterase inhibitors) is usually allowed The utility of structural MRI as a biomarker
and therapeutic efficacy in Alzheimer’s dis- as background medication in Alzheimer’s dis- in clinical trials will thus be increased by stan-
ease.”18. However, despite data showing ease clinical trials. Therefore, it is important dardization of acquisition and analysis meth-
that the drug, even at high doses, does not that theragnostic biomarkers used to monitor ods12. A task force to develop a harmonized
affect CSF Aβ42 concentrations or cause a the effect of an anti-Aβ drug are not affected procedure for evaluation of hippocampal
shift toward shorter Aβ isoforms in CSF in by this type of treatment. Several studies atrophy is ongoing under the auspices of the
humans19, and a phase 2 trial showing no have shown that the CSF biomarkers Aβ42, Alzheimer’s Association.
overall effect on primary outcome measures T-tau and P-tau do not change during cho- Similar standardization issues also apply
in individuals with Alzheimer’s disease20, linesterase inhibitor treatment and also have to PET imaging. For amyloid PET, there is
the drug was taken to a large phase 3 clinical a very low intraindividual variability, both in no consensus on the optimal brain region
trial. This 18-month trial on more than 1,600 short-term and long-term trials25,26. These to quantify amyloid retention and very lim-
subjects with Alzheimer’s disease showed no results indicate that even minor changes in ited data comparing the performance of the
significant effects on cognitive or functional biomarker amounts can be monitored with various radioligands (PiB, FDDNP, AV-45 and
primary outcomes21. CSF biomarkers. It is noteworthy that stud- AZD2184)13. For FDG-PET, further data is
Biomarkers used to detect and monitor ies have shown that treatment with donepezil needed on which brain regions to evaluate in
the biochemical effects of therapeutics may may retard both the decline in brain glucose each stage of the disease, that is, hypometabo-
be called ‘theragnostic’ biomarkers22. These metabolism evaluated by FDG-PET27,28 and lism in the parietotemporal cortex to diagnose
types of biomarkers may be valuable in drug the progression rate of hippocampal atrophy Alzheimer’s disease, in the medial temporal
development to bridge the gap between animal measured by MRI29,30. Thus, cholinesterase lobe to predict MCI and in the posterior cin-
studies and large clinical trials. Biomarker evi- treatment might be a potential confounder gulate cortex to predict progression from MCI
dence from small-scale trials that a drug has a when evaluating the effect of disease-modify- to Alzheimer’s disease13.
true effect on the Alzheimer’s disease process ing therapies with these types of biomarkers.
directly in humans would help in selecting Implementing theragnostic biomarkers
the most promising drug candidates, thereby Unresolved issues for Alzheimer’s in clinical trials
improving the success rate of large clinical disease biomarkers Conceptually, it may be useful to divide bio-
phase 2 and 3 trials. Such trials could be short- Both imaging and CSF biomarkers show markers into primary (specific) and second-
term proof-of-principle studies on a limited promise as diagnostic and theragnostic tools ary (downstream) biomarkers (Fig. 1). A
number of healthy volunteers in phase 1 in clinical trials, but they have in common a primary biomarker can be used to identify
(ref. 23), as well as proof-of-concept studies need for standardization. Although biological and monitor the specific biochemical effect,
on individuals with Alzheimer’s disease in and within-laboratory variability for CSF bio- or mode of action, of a drug. In trials with
phase 2 (ref. 24). This type of early clinical markers is low, there is a variation in biomarker Aβ-targeting compounds such as secretase
biomarker study would aid decision making levels between laboratories. Furthermore, there inhibitors or Aβ immunotherapy, examples
regarding whether to embark on large and are no available calibrators (certified reference of primary biomarkers include CSF Aβ42 and
expensive phase 3 trials. standards) for these biomarkers and no consen- Aβ40, β-secretase (BACE1) activity and sol-
sus on what assays should be used. This com- uble APP-β and amyloid-PET. Downstream
What constitutes a proficient plicates multicenter trials and also precludes biomarkers are used to identify and monitor
theragnostic biomarker? the introduction of generally applicable cutoff effects on pathogenic processes downstream
A theragnostic biomarker should reflect levels. In response to this problem, a global of the drug target, for example, CSF T-tau
the central pathogenic processes of the dis- quality-control program for CSF biomark- and MRI measurements of brain atrophy to
ease. In contrast to drug development for ers, supported by the Alzheimer’s Association, identify and monitor an effect on the rate of
other neurodegenerative disorders, such as has recently been launched22. The aim of this neuronal degeneration in an anti-Aβ trial.
Parkinson’s disease, Alzheimer’s disease drug program is to serve as the basis for efforts to A key question will also be which of the
development has the benefit of several such minimize variation caused by differences in biomarkers to include in a trial. For example,
biomarkers readily available. CSF concentra- preanalytical and laboratory procedures. A several studies have shown a strong correla-
tions of T-tau reflect cortical axonal degenera- substantial part of the variability is probably tion between low CSF Aβ42 concentration
tion, whereas P-tau reflects tangle pathology also caused by assay-related factors, espe- and high binding of the PiB ligand on PET,
and Aβ42 reflects brain amyloid pathology22. cially batch-to-batch variation of assays. This suggesting that these biomarkers will give
Further, MRI imaging of hippocampal atro- involves a need for assay vendors to implement similar information on the amount of Aβ
phy gauges progression of neurodegenera- improved standards for quality control in assay plaque pathology31,32. For a clinical trial, fac-
nature medicine advance online publication 3
commentary
tors such as the willingness among clinicians duced by a previously unknown pathway of in CSF, suggesting that the treatment may have
to perform lumbar puncture on the one hand, APP processing38. A series of studies in dogs, reduced the rate of neuronal degeneration44.
and the availability of cyclotrons and PET monkeys and Alzheimer’s disease–transgenic For MRI measurements of atrophy, a decrease
scanners and financial consideration on the mice have shown that γ-secretase inhibitor in atrophy rate would be the logical expecta-
other, will form a basis for a decision on which treatment results in a marked increase in tion for a drug with a disease-modifying
of these biomarkers to choose. Aβ1–14, Aβ1–15 and Aβ1–16 abundance, effect. However, the AN1792 trial showed an
probably as a result of increased substrate unexpected increase in the brain atrophy rate
Can we find new biomarkers? availability of the C99 APP stub induced by in antibody responders, possibly as a result
A surrogate biomarker can be used as a sub- γ-secretase inhibition39–41. A recent clinical of removal of Aβ plaques45. A recent report
stitute for a clinical end point in a clinical trial on the γ-secretase inhibitor LY450139 on the bapineuzumab immunotherapy trial
trial33. In the case of Alzheimer’s disease, such in Alzheimer’s disease found no effect on the showed a reduced cortical PiB retention during
a biomarker should correlate with cognitive CSF concentrations of Aβ1–40 or Aβ1–42, as treatment compared with both baseline and
or functional clinical outcomes. Synapses measured by ELISA42. In another study in the placebo17. These results bring hope that amy-
are the primary functional unit for neuronal same trial, a marked dose-dependent increase loid PET will be valuable to assess the effect of
communication, and the degree of synaptic in Aβ1–14, Aβ1–15 and Aβ1–16 was found by Aβ-targeting disease-modifying therapies on
degeneration has also been found to show the the immunoprecipitation–MALDI-TOF tech- cortical fibrillar Aβ load in clinical trials.
best correlation with severity of dementia in nique24. Thus, these short Aβ isoforms may be Thus, there are only limited data from
Alzheimer’s disease34. Synaptic proteins might valuable and sensitive biomarkers to monitor Alzheimer’s disease trials suggesting that bio-
thus serve as valuable surrogate CSF biomark- the biochemical effect of γ-secretase inhibitor markers have a place as theragnostic markers
ers, as the abundance of these molecules is treatment in clinical trials. in phase 1–2 studies or surrogate end points
likely to correlate with cognitive function and in phase 3 studies. A challenge in this field of
disease progression. Although synaptic pro- Use of biomarkers to label a drug research is the lack of drugs with an estab-
teins, such as synaptotagmin and rab3a, have ‘disease modifying’ lished disease-modifying effect that can be
been shown to be present in human CSF35, Ideally, a change in the slope of cognitive used to validate a biomarker and, at the same
assay development for this class of proteins decline may identify differences between time, that biomarker evidence that a drug
has been difficult, owing to their low concen- symptomatic and disease-modifying drugs. candidate affects the central disease processes
trations in CSF. Clinical trials designed with a delayed start will be required to label a drug as disease
Probably for the same reason, assay devel- (placing patients on drug or placebo for a modifying33. This means that Alzheimer’s
opment has been difficult for Aβ oligomers period at the start of the trial and later ini- disease drug development and biomarker
in CSF. Aβ oligomers would be a valuable tiating treatment also in the placebo group) research need to go forward hand in hand.
biomarker in Alzheimer’s disease drug or staggered withdrawal (placing patients on The increasing number of clinical trials on
development for several reasons. First, recent active drug at the trial start and later random- disease-modifying drug candidates that
experimental data suggest that soluble Aβ oli- izing them to drug or placebo) have been include biomarkers for enrichment of pure
gomers inhibit long-term potentiation and suggested to allow this differentiation. This Alzheimer’s disease cases or as end points
have synaptotoxic properties, and thereby approach may be theoretically attractive, but it will hopefully keep providing accumulative
have a central role in Alzheimer’s disease has not been successful, probably owing to the evidence for the usefulness of biomarkers in
pathogenesis36. Second, it is difficult to pre- very large variability in the rate of cognitive Alzheimer’s disease clinical trials and serve
dict in which direction CSF Aβ42 may change change between cases, both in the dementia as the basis for approval by the regulatory
in an Aβ immunotherapy trial. Thus, CSF Aβ and the MCI stages of Alzheimer’s disease. authorities of disease-modifying drugs for
oligomers might be an important biomarker Academic institutions, the pharmaceuti- this devastating disease.
for Alzheimer’s disease. Some preliminary cal industry and regulatory organizations
COMPETING FINANCIAL INTERESTS
studies on Aβ oligomers in CSF have been agree that biomarkers have a crucial role in
The author declares competing financial interests:
published; a recent study with a previously the drug development process and that evi- details accompany the full-text HTML version of the
undescribed ELISA assay found a clear dence obtained from biomarker studies show- paper at http://www.nature.com/naturemedicine/.
increase in CSF Aβ oligomers in Alzheimer’s ing that a drug candidate affects the central
1. Blennow, K., de Leon, M.J. & Zetterberg, H. Lancet
disease37. disease processes in Alzheimer’s disease will, 368, 387–403 (2006).
Targeted proteomics studies have shown together with a beneficial effect on cognition, 2. Hardy, J. & Selkoe, D.J. Science 297, 353–356
that, in addition to Aβ1–42 and Aβ1–40, there be essential for a drug to be labeled disease (2002).
3. Mangialasche, F. et al. Lancet Neurol. 9, 702–716
are several shorter Aβ isoforms truncated at modifying33. (2010).
the carboxy terminus. By a technique based 4. Holmes, C. et al. Lancet 372, 216–223 (2008).
on immunoprecipitation with an Aβ-specific How far have we come today? 5. Garcia-Alloza, M. et al. Mol. Neurodegener. 4, 19
(2009).
monoclonal antibody combined with matrix- Apart from animal studies, there is only pre- 6. DeCarli, C. Lancet Neurol. 2, 15–21 (2003).
assisted laser desorption–ionization time-of- liminary evidence that biomarkers may be 7. Petersen, R.C. et al. N. Engl. J. Med. 352, 2379–2388
(2005).
flight (MALDI-TOF) mass spectrometry, all useful as theragnostic markers in humans.
8. Hansson, O. et al. Lancet Neurol. 5, 228–234
Aβ isoforms can be quantified simultane- For CSF biomarkers, a phase 2 study of the Aβ (2006).
ously38. Cell culture studies have shown that clearance-enhancing compound PBT2 showed 9. Shaw, L.M. et al. Ann. Neurol. 65, 403–413 (2009).
10. Visser, P.J. et al. Lancet Neurol. 8, 619–627 (2009).
the longer isoforms (Aβ1–17 up to Aβ1–42) a dose-dependent reduction in the amount 11. Mattsson, N. et al. J. Am. Med. Assoc. 302, 385–393
are produced by the amyloidogenic pathway, of the primary biomarker Aβ42 in CSF43. (2009).
by the concerted action of β-secretase and Furthermore, in the AN1792 immunother- 12. Frisoni, G.B., Fox, N.C., Jack, C.R. Jr., Scheltens, P. &
Thompson, P.M. Nat. Rev. Neurol. 6, 67–77 (2010).
γ-secretase, whereas the short Aβ isoforms apy trial, there was a decrease toward normal 13. Nordberg, A., Rinne, J.O., Kadir, A. & Langstrom, B.
(Aβ1–14, Aβ1–15 and Aβ1–16) are pro- amounts of the downstream biomarker T-tau Nat. Rev. Neurol. 6, 78–87 (2010).
4 advance online publication nature medicine
commentary
14. Nelson, P.T., Kukull, W.A. & Frosch, M.P. J. Neuropathol. 25. Blennow, K. et al. Neurosci. Lett. 419, 18–22 35. Davidsson, P., Puchades, M. & Blennow, K.
Exp. Neurol. 69, 449–454 (2010). (2007). Electrophoresis 20, 431–437 (1999).
15. Kotzbauer, P.T., Trojanowsk, J.Q. & Lee, V.M. J. Mol. 26. Zetterberg, H. et al. J. Alzheimers Dis. 12, 255–260 36. Walsh, D.M. & Selkoe, D.J. J. Neurochem. 101, 1172–
Neurosci. 17, 225–232 (2001). (2007). 1184 (2007).
16. Schneider, J.A., Arvanitakis, Z., Leurgans, S.E. & 27. Chen, X., Magnotta, V.A., Duff, K., Boles Ponto, L.L. 37. Fukumoto, H. et al. FASEB J. published online,
Bennett, D.A. Ann. Neurol. 66, 200–208 (2009). & Schultz, S.K. J. Neuropsychiatry Clin. Neurosci. 18, doi:10.1096/fj.09-150359 (25 March 2010).
17. Rinne, J.O. et al. Lancet Neurol. 9, 363–372 (2010). 178–185 (2006). 38. Portelius, E. et al. Neurobiol. Aging published online,
18. Eriksen, J.L. et al. J. Clin. Invest. 112, 440–449 28. Tune, L. et al. Am. J. Geriatr. Psychiatry 11, 169–177 doi:10.1016/j.neurobiolaging.2009.06.002 (14 July
(2003). (2003). 2009).
19. Galasko, D.R. et al. Alzheimer Dis. Assoc. Disord. 21, 29. Krishnan, K.R. et al. Am. J. Psychiatry 160, 2003– 39. Portelius, E. et al. Neurodegener. Dis. 6, 258–262
292–299 (2007). 2011 (2003). (2009).
20. Wilcock, G.K. et al. Lancet Neurol. 7, 483–493 30. Hashimoto, M. et al. Am. J. Psychiatry 162, 676–682 40. Cook, J.J. et al. J. Neurosci. 30, 6743–6750 (2010).
(2008). (2005). 41. Portelius, E. et al. J. Alzheimers Dis. published online,
21. Green, R.C. et al. J. Am. Med. Assoc. 302, 2557–2564 31. Fagan, A.M. et al. Ann. Neurol. 59, 512–519 (2006). doi:10.3233/JAlzheimer’s disease-2010-100573 (15
(2009). 32. Degerman Gunnarsson, M. et al. Dement. Geriatr. Cogn. July 2010).
22. Blennow, K., Hampel, H., Weiner, M. & Zetterberg, H. Disord. 29, 204–212 (2010). 42. Fleisher, A.S. et al. Arch. Neurol. 65, 1031–1038
Nat. Rev. Neurol. 6, 131–144 (2010). 33. Hampel, H. et al. Nat. Rev. Drug Discov. 9, 560–574 (2008).
23. Bateman, R.J. et al. Ann. Neurol. 66, 48–54 (2009). (2010). 43. Lannfelt, L. et al. Lancet Neurol. 7, 779–786 (2008).
24. Portelius, E. et al. Alzheimers. Res. Ther. 2, 7 34. Davidsson, P. & Blennow, K. Int. Pychogeriatr. 10, 44. Gilman, S. et al. Neurology 64, 1553–1562 (2005).
(2010). 11–23 (1998). 45. Fox, N.C. et al. Neurology 64, 1563–1572 (2005).
nature medicine advance online publication 5
C O M M E N TA RY
Clinical trials of disease-modifying
therapies for neurodegenerative diseases:
the challenges and the future
Anthony E Lang
Neurodegenerative diseases such as provided by families and others, estimated course of neurodegenerative diseases, I will
Parkinson’s disease and Alzheimer’s to have been $144 billion in 2009 (ref. 2). largely restrict my comments to small mol-
disease represent a crucial and In the UK, the cost of dementia is now esti- ecule treatments directed at the underlying
exponentially increasing challenge to mated to exceed the combined cost of cancer, neurodegenerative processes, but many of the
health care systems throughout the heart disease and stroke3. The prevalence of important issues apply broadly to all of these
world. There is an urgent need for Parkinson’s disease is expected to double by types of therapies.
effective treatments that will both delay the year 2030 (ref. 4), and, given the greater Although the term ‘neuroprotection’ has
their onset and slow their inexorable risk of Parkinson’s disease dementia in been used widely with respect to early clinical
progression. Many obstacles stand elderly individuals with Parkinson’s disease, trials in Parkinson’s disease and Alzheimer’s
in the way of realizing these goals. it is expected that the health care costs for disease7,8, a positive result in such a clinical
It is expected that future advances this disorder will increase even more rela- trial does not mean that the drug is acting on
will have a major impact on how and tive to the prevalence of the disease. These the underlying biological mechanisms that
when the diagnosis will be made. It is concerns highlight the urgent need for the cause the disease. Although the future ideal
hoped that these will eventually make development of effective disease-modifying may be to confirm true neuroprotection, in
it possible to initiate effective disease- therapies. Effective treatment would have a the immediate future the best we can hope
modifying therapies long before the major influence on the economic and social for is disease modification, and, if we succeed
neurodegenerative process becomes burden of these age-related disorders. For in truly altering the inexorably progressive
established and symptomatic. example, in the case of Alzheimer’s disease, clinical course of these diseases, it shouldn’t
it has been estimated that a delay in onset by matter how this is accomplished. Presentation
Neurodegenerative diseases of the elderly 5 years would translate into a 50% decrease with signs and symptoms of these disorders
currently represent a major challenge to the in disease prevalence, and a delay of 10 years probably represents a combination of the
health care system. With the increasing lon- would result in a virtual disappearance of the disease reaching a critical threshold of neu-
gevity of the general population, the predicted disease5,6. Unfortunately, despite considerable ronal dysfunction along with the failure of a
figures for the prevalence of these disorders investment, to date all attempts at developing number of central nervous system compen-
and their global financial impact are truly such treatments have failed. satory mechanisms that have been activated
staggering. For example, it has been estimated in response to the underlying neurodegen-
that by 2030 as many as 7.7 million people in Terminology eration. For example, it is well known that
the US will have Alzheimer’s disease, and by The ‘Holy Grail’ of neurodegenerative dis- individuals with Parkinson’s disease have pro-
2050 this number will reach approximately eases is the development of effective ‘neuro- found nigrostriatal dopamine cell loss before
13.5 million, with total annual costs for care protective’ therapy. By definition, this implies developing clinical motor features9 (this is
rising from $172 billion in 2010 to $1.08 tril- that the treatment has a direct impact on the particularly extreme in people with auto-
lion in 2050 (ref. 1). This analysis does not biology of the disease, as confirmed by a somal recessive forms of Parkinsonism, such
take into account the value of unpaid care reliable measure of this effect. Alternatively, as those with Parkin mutations10). Their final
‘disease modification’ implies a modification presentation with motor symptoms probably
Anthony E. Lang is in the Department of Medicine of the clinical course of the disease without represents a failure of a variety of compensa-
(Neurology) at the University of Toronto, Toronto, implicating mechanism. Other interventions tory mechanisms. If it were possible to even
Canada. could be classified as ‘neurorestorative’ (for partially restore some of these mechanisms
e-mail: lang@uhnresearch.ca example, cell-based and some gene thera- before the individual reaches an irretriev-
pies) and ‘neuroregenerative’ (for example, able degree of degeneration, the clinical
Published online 21 September 2010; trophic factors). Although these experimen- course of the disease might be considerably
doi: 10.1038/nm.2220 tal approaches hold promise for changing the altered without even changing the underly-
nature medicine advance online publication 1
commentary
ing neurodegenerative process. Being able Hypothetical course
of neuronal loss
to identify people at the appropriate point in
their disease pathogenesis would, therefore, Mildly effective drug
Progressive neuronal degeneration
be extremely important. Moderately effective drug
Problems in planning, designing and
conducting clinical trials 3
One of the obvious challenges to changing the
course of neurodegenerative diseases relates
Threshold for
to our incomplete or inadequate understand- premotor features
ing of the underlying disease pathogeneses.
Age remains the single most important risk 2
Threshold for
factor for Alzheimer’s disease and Parkinson’s classical motor features
disease11, yet its influence on the underlying 1
biological mechanisms is not understood.
Particularly in Parkinson’s disease, age also
has a strong influence on clinical manifesta-
tions (that is, there is greater cognitive and Time Premotor period Overt clinical symptoms
axial motor disturbances in those with later Figure 1 Theoretical effects of disease modifying treatments in Parkinson’s disease. Effects of
age of onset)12,13, and this will have a major initiation of two disease-modifying treatments at different stages of Parkinson’s disease on the clinical
impact on many aspects of clinical trial features and course of the illness. 1, after onset of classical clinical features of the disease; 2, after the
onset of premotor features (before classical motor); 3, before the onset of any clinical features in at-risk
design, including subject selection and treat-
individuals or with the use of a sensitive disease trait biomarker.
ment outcomes.
To date, animal models for both Alzheimer’s
disease and Parkinson’s disease have failed to vidual molecular targets, this may require Parkinson Disease (LABS-PD), directed by the
reflect many of the key aspects of the human innovative drug discovery strategies that fall Parkinson Study Group26.
diseases14–19. Such models have been espe- outside the current single-target approach A preeminent goal of these and other studies
cially ineffective in predicting the response that is the paradigm within the pharmaceuti- is the development of various types of biomark-
of affected individuals to putative disease- cal industry. Clearly, advances in this regard ers that will be essential components in realiz-
modifying therapies20. If more reliable, more will require preclinical animal models that are ing effective disease-modifying therapies27–29.
representative animal models can be devel- more translatable to the human disease than Markers that accurately track the progression
oped, success in establishing effective disease- the current ones and that can be well character- of the underlying disease (progression marker)
modifying therapies for humans will require ized by multimodal micro–positron emission and that can serve as surrogates of the desired
robust and easily replicated effects, preferably tomography (PET) and functional magnetic therapeutic effect (response marker) will be
in several animal species21. resonance imaging procedures. crucial for use in phase 2 clinical trials allow-
Therapeutic approaches that are directed at Neurodegenerative diseases such as ing prioritization of compounds for subsequent
single biological mechanisms or targets may Alzheimer’s disease and Parkinson’s disease study in phase 3 trials. One major stumbling
be inadequate, given the complexity of these are quite heterogeneous at multiple levels, block to the development of disease-modifying
multifaceted neurodegenerative disorders. including their genetic, clinical and neuro- therapy is the inability to demonstrate and mea-
Successful treatment may require a cocktail pathological bases, so disease pathogenesis is sure the central effects of the study drug, which
approach, such as that applied in cancer. These probably equally heterogeneous. This hetero- also compromises the selection of drug dos-
treatments may vary depending on the stage of geneity may therefore translate into a variety age in clinical trials. Pharmacodynamic mark-
the disorder being treated. For example, very of responses to putative disease-modifying ers would allow the direct measurement of the
early treatment in at-risk individuals might be drugs in each patient subgroup. Despite wide- biological effects on the proposed drug targets,
exclusively directed at selected primary disease spread recognition of this important feature of thus supporting drug development decisions.
mechanisms, whereas later treatment may need neurodegenerative diseases, to date, treatment ‘Theragnostic markers’ is another term used to
to address mechanisms that are further down- trials have largely lumped all patients together describe biomarkers designed to identify and
stream and additional secondary mechanisms, in the expectation of a uniform therapeu- monitor the biochemical effect of therapeutic
including neuroinflammation22. Furthermore, tic response. However, a number of ongoing interventions30. Trials employing such a bio-
we have absolutely no understanding of the large-scale studies designed to evaluate clini- marker could involve a relatively small num-
basis of the selective vulnerability of various cal, imaging and other biological markers and ber of patients for short treatment periods, thus
neuronal populations in neurodegenerative their natural histories promise to have a major allowing faster decisions of whether to proceed
diseases. A better understanding of how and impact on the understanding of this hetero- with large and expensive phase 2 or 3 clinical tri-
why certain regions of the brain are more geneity and therefore on the design of future als. A diagnostic marker would improve accu-
affected than others (for example, through trials of disease-modifying therapies. These racy in diagnosing patients that enter phase 3
sharing of common biological functions or include the Alzheimer’s Disease Neuroimaging trials, increasing the power to detect disease
being connected in common neuronal net- Initiative in the US and similar projects in modification and reducing costs. A practical
works) may assist in the choice of agents to other countries23–25 and the Michael J. Fox example of the need for this type of biomarker
include in future neuroprotective cocktails. Foundation–supported Parkinson Progression is already well recognized from experience in
Additionally, if therapeutic efficacy requires Marker Initiative (PPMI), as well as the pro- trials in early Parkinson’s disease, in which a
modulating neural networks rather than indi- posed Longitudinal and Biomarker Studies in proportion of enrolled subjects did not have
2 advance online publication nature medicine
commentary
the disease of interest—presynaptic dopamine type, prodromal Alzheimer’s dementia and treatment31,45,46. However, although imaging
imaging has yielded scans without evidence of preclinical Alzheimer’s disease are under active of the status of the presynaptic nigrostriatal
dopaminergic deficit (SWEDD) in up to 14% consideration. Likewise, studies evaluating var- dopamine system can readily distinguish indi-
of study participants31. Diagnostic markers ious ‘premotor’ features of Parkinson’s disease viduals with early Parkinson’s disease from con-
may also be crucial in dividing patients into (for example, olfactory dysfunction, rapid eye trols, and abnormalities can be observed even
subgroups according to who will be more or movement behavior disorder and changes in before motor symptoms and signs are appar-
less responsive to therapies directed at specific heart rate variability) are being combined with ent, these studies have been disappointing in
disease-related targets. This would reduce the central nervous system imaging (for example, monitoring both disease-modifying34 and
heterogeneity of enrolled subjects and increase dopamine transporter single-photon emis- neurorestorative47,48 therapies. This may be
the likelihood of demonstrating specific target– sion computed tomography and transcranial due, in part, to the potential pharmacological
related impact in the trial. Pharmacogenomic ultrasound) in an attempt to define the pres- modulation or regulation of presynaptic pro-
markers that predict serious adverse medica- ence of the disease before the development of teins that may not relate to the actual disease
tion effects would also assist greatly in early the typical motor features33–35. In planning status. Fluorodeoxyglucose (FDG)-PET with
drug development. disease-modifying trials in at-risk individuals multivariate principal components analysis
Finally, biomarkers allowing a definitive or in those with very early preclinical disease shows promise in providing an alternative
diagnosis at the earliest stages of disease rep- (this term may not be truly accurate, as many method of monitoring disease status. However,
resent one of the highest priorities in the search affected individuals will have clinical features experience with this technique in Parkinson’s
for effective disease-modifying therapies. The that simply don’t constitute the diagnosis as disease is largely limited to one center49,50.
goal is to be able to diagnose patients before the currently conceived), establishing dependable In the absence of reliable biomarkers of dis-
establishment of widespread neuronal damage, primary endpoints will be problematic in the ease state that are not regulated by the drug that
at a time when they would presumably be more absence of a reliable biomarker of disease state. is being tested, any assessment of disease modi-
likely to respond to the effects of the therapeu- One common consideration is the conversion fication becomes extremely difficult once effec-
tic intervention. Depending on the safety and to manifest disease, but establishing an early tive symptomatic treatment is initiated. The
cost of the treatment, an alternative would and accurate clinical diagnosis of manifest best example of this is the use of dopaminergic
be to identify individuals with a high risk of Parkinson’s disease or Alzheimer’s disease may medication for Parkinson’s disease; once this
developing neurodegeneration even before be methodologically challenging36. has been started, clinical assessments become
it has begun, such as individuals inheriting extremely limited in their ability to reflect
various disease-associated genetic mutations Study design issues disease progression. Therefore, long-term
or genetic risk factors. A widespread concern To date, neuroprotective trials have largely studies in patients who are already on these
related to the large trials conducted to date has evaluated people in the early clinical stages of dopaminergic treatments are now emphasizing
been that individuals with well-established the disease (generally untreated), using clini- features that tend to be resistant to dopamin-
diagnoses, even in the earliest clinical stages of cal endpoints that involve either the change in ergic medication (so-called nondopaminergic
the disease, may already suffer from a neuro- a classical clinical measure of the disease over features, believed to be due to the extension of
degenerative process that is too far advanced to time or progression to the point of reaching a the disease beyond the nigrostriatal system51).
benefit from treatment, particularly from the disease milestone (for example, need for dop- These include ambulatory capacity (freezing,
more focused treatments directed at a single aminergic therapy in Parkinson’s disease tri- falling, gait abnormalities and postural sta-
pathogenetic mechanism. An analogy might als37–39). The greatest concern in these studies bility), cognitive dysfunction, disability and
be that of ischemic heart disease, where early has been the potential for the study interven- quality of life. New statistical approaches to
treatment of various risk factors such as hyper- tion to cause symptomatic benefit that pre- assess these outcomes are also required. For
tension, hypercholesterolemia, coronary artery cludes the determination of disease-modifying example, in the Neuroprotection Exploratory
narrowing and so on may be quite effective but, effects. Studies using a washout design, where Trials in Parkinson’s Disease (NET-PD) Long-
once congestive heart failure is established, the drug is removed after a certain period of term Study 1 comparing creatine to placebo,
treating these factors may be too late to affect treatment, have failed to adequately overcome five measures of decline will be combined into
the outcome. this problem, as the symptomatic treatment a single outcome and compared by a nonpara-
As our understanding of neurodegenerative effect may exceed the duration of the washout. metric Global Statistical Test7. This novel sta-
diseases progresses, it is clear that there will The delayed-start design40,41 was developed tistical approach is especially well suited to a
be a need to diagnose these disorders at the in hopes of overcoming this confounding multidimensional disorder such as Parkinson’s
very earliest stages. Our definitions of these effect. In such a study, an initial placebo con- disease where patient disability and functional
diseases will necessarily change from requir- trol phase is followed by a second phase in impairment are due to a combination of
ing early cognitive or motor disturbances in which all patients receive the active treatment. disease- and treatment-related disturbances in
Alzheimer’s and Parkinson’s, respectively, to However, such an approach is not without its diverse brain systems52.
considering even earlier clinical, and preferably own potential problems42,43. Although simula- The lack of biomarkers also hampers the
biological, disturbances. This would allow the tion studies have shown a clear advantage of choice of which promising disease-modifying
initiation of treatments that might completely washout versus delayed-start designs44, the treatments to pursue. One approach taken by
prevent the development of the clinical fea- prolonged washouts necessary may not be the NET-PD program has been to evaluate
tures currently required for diagnosis (Fig. 1). practical or possible in the case of treatments a variety of agents through a futility trial
Proposals already exist for the development of that provide substantial and needed symptom- design, as originally applied in oncology53.
new diagnostic criteria that include biomark- atic benefit. The rate of decline of a neuroim- This approach attempts to quickly screen
ers for the early diagnosis of Alzheimer’s dis- aging surrogate has been used in Parkinson’s out agents associated with a disease course
ease32, and additional diagnostic categories of disease in the hopes of avoiding the confound- not sufficiently different from historical
mild cognitive impairment of the Alzheimer’s ing effects of clinical symptomatic benefit of controls. Those compounds not rejected as
nature medicine advance online publication 3
commentary
futile can then proceed to further study in a (PPMI, LABS-PD) similar to those applied to 340–347 (2009).
19. Philipson, O. et al. FEBS J. 277, 1389–1409
long-term ‘large simple’ trial design. In this Alzheimer’s disease over the past 5 years will (2010).
type of study, individuals with midstage dis- set the stage for more successful study of puta- 20. Lang, A.E. Lancet Neurol. 5, 990–991 (2006).
ease are randomly assigned to receive active tive disease-modifying treatments. 21. Jucker, M. Nat. Med. published online, doi:10.1038/
nm.2224 (21 September 2010)
study drug or placebo and are followed for As advances are made on the fronts high- 22. Hirsch, E.C. & Hunot, S. Lancet Neurol. 8, 382–397
long periods (5–10 years) during which they lighted above, it is expected that major changes (2009).
23. Aisen, P.S. et al. Alzheimers Dement. 6, 239–246
are treated with routine symptomatic therapy will be possible in the design of studies evalu- (2010).
according to the judgment of the investiga- ating potential disease-modifying therapies. 24. Trojanowski, J.Q. et al. Alzheimers Dement. 6, 230–
tor. An interesting sideline challenge to this Such crucial components of clinical trials as 238 (2010).
25. Weiner, M.W. et al. Alzheimers Dement. 6, 202–211
approach in Parkinson’s disease has been the the primary inclusion criteria, the number of (2010).
observation that historical controls may not subjects required to determine efficacy and the 26. Ravina, B. et al. Mov. Disord. 24, 2081–2090
adequately establish futility threshold, owing primary outcome variables will be very differ- (2009).
27. Scherzer, C.R. Neurobiol. Dis. 35, 148–156 (2009).
to possible changes over the years in practice ent from those used currently. For approval 28. Hampel, H. et al. Nat. Rev. Drug Discov. 9, 560–574
parameters, in the use of the clinical rating of a treatment with an indication of disease (2010).
29. Blennow, K. Nat. Med. published online, doi:10.1038/
scale (the Unified Parkinson’s Disease Rating modification, it is likely that, initially, combi- nm.2221 (21 September 2010).
Scale) or in poorly understood changes in the nations of clinical and biomarker outcomes 30. Blennow, K., Hampel, H., Weiner, M. & Zetterberg, H.
patient population of interest53. Alternative will be required. However, as more accurate Nat. Rev. Neurol. 6, 131–144 (2010).
31. Fahn, S. et al. N. Engl. J. Med. 351, 2498–2508
study designs might also be considered in the and reliable biomarkers become available, there (2004).
early evaluation of possible disease-modify- will hopefully come a time that outcomes will 32. Dubois, B. et al. Lancet Neurol. 6, 734–746 (2007).
ing treatments. For example, where rates of rely exclusively on nonclinical evaluations, and 33. Siderowf, A. & Stern, M.B. Ann. Neurol. 64,
S139–S147 (2008).
conversion or changes in outcomes are not successful treatment will completely prevent the 34. Ponsen, M.M., Stoffers, D., Twisk, J.W.R., Wolters,
well known in advance, adaptive designs onset of the clinical disorders we now recognize E.C. & Berendse, H.W. Mov. Disord. 24, 1060–1065
assist in drug development by introducing as Alzheimer’s disease and Parkinson’s disease. (2009).
35. Haehner, A. et al. Mov. Disord. 22, 839–842
flexibility within trial design, allowing pre- (2007).
ACKNOWLEDGMENTS
specified modifications in a seamless fashion 36. Kieburtz, K. Neurology 66, S50–S57 (2006).
I thank B. Greenberg for his helpful comments. 37. Parkinson Study Group. N. Engl. J. Med. 321, 1364–
on the basis of ongoing accruing data (http://
1371 (1989).
biopharmnet.com/doc/doc12004.html)54. COMPETING FINANCIAL INTERESTS 38. Parkinson Study Group PRECEPT Investigators et al.
Currently, it must be emphasized that The author declares no competing financial interests. Neurology 69, 1480–1490 (2007)
39. Olanow, C.W. et al. Lancet Neurol. 5, 1013–1020
Alzheimer’s disease and Parkinson’s dis- (2006).
ease differ greatly in their prospects and in 1. The Lewin Group. Saving lives, saving money: dividends
40. Leber, P. Alzheimer Dis. Assoc. Disord. 11 Suppl 5,
for Americans investing in Alzheimer’s Research. (Falls
approaches to planning disease-modifying Church, Virginia, USA, 2004).
S10–S21 (1997).
41. Olanow, C.W. et al. Mov. Disord. 23, 2194–2201
therapy. Alzheimer’s disease is considerably 2. Alzheimer’s Association. <http://www.alz.org/
(2008).
documents_custom/trajectory.pdf> (2010).
further along than Parkinson’s disease with 3. Alzheimer’s Disease International. <http://www.alz.
42. Clarke, C.E. Mov. Disord. 23, 784–789 (2008).
respect to nonclinical criteria that might be 43. D’Agostino, R.B. Sr. N. Engl. J. Med. 361, 1304–
co.uk/research/files/WorldAlzheimerReport.pdf>
1306 (2009).
used in the evaluation of new treatments. (2009).
44. Ploeger, B.A. & Holford, N.H. Pharm. Stat. 8, 225–
4. Dorsey, E.R. et al. Neurology 68, 384–386 (2007).
These include cerebrospinal fluid markers of 5. DeKosky, S.T. & Marek, K. Science 302, 830–834 238 (2009).
different components of the disease (such as (2003). 45. Parkinson Study Group et al. J. Am. Med. Assoc. 287,
6. Brookmeyer, R., Gray, S. & Kawas, C. Am. J. Public 1653–1661 (2002).
Aβ42 or tau), imaging of the underlying bio- 46. Whone, A.L. et al. Ann. Neurol. 54, 93–101 (2003).
Health 88, 1337–1342 (1998).
logical changes (amyloid imaging) and pro- 7. Olanow, C.W., Kieburtz, K. & Schapira, A.H. Ann. 47. Freed, C.R. et al. N. Engl. J. Med. 344, 710–719
(2001).
gressive metabolic and anatomic correlates Neurol. 64 Suppl 2, S101–S110 (2008).
48. Olanow, C.W. et al. Ann. Neurol. 54, 403–414
8. Francis, P.T., Norberg, A. & Arnold, S.E. Trends
of the status of the underlying neurodegen- Pharmacol. Sci. 26, 104–111 (2005). (2003).
eration (FDG-PET and volumetric magnetic 9. Brooks, D.J. Ann. Neurol. 44, S10–S18 (1998). 49. Tang, C.C., Poston, K.L., Dhawan, V. & Eidelberg, D.
J. Neurosci. 30, 1049–1056 (2010).
resonance imaging, respectively)29. The addi- 10. Broussolle, E. et al. Neurology 55, 877–879 (2000).
50. Eckert, T., Tong, C. & Eidelberg, D. Lancet Neurol. 6,
11. Levy, G. Arch. Neurol. 64, 1242–1246 (2007).
tion of a genetic risk factor (apolipoprotein E 12. Levy, G. et al. Arch. Neurol. 62, 467–472 (2005). 926–932 (2007).
ε4 status) can also be included; indeed, it has 13. Aarsland, D. et al. J. Neurol. 254, 38–45 (2007). 51. Lim, S.Y., Fox, S.H. & Lang, A.E. Arch. Neurol. 66,
14. Jenner, P. Ann. Neurol. 64, S16–S29 (2008). 167–172 (2009).
already been found that the effectiveness and 52. Huang, P. et al. Mov. Disord 24, 1732–1739
15. Dawson, T.M., Ko, H.S. & Dawson, V.L. Neuron 66,
adverse event profile of one form of passive 646–661 (2010). (2009).
Aβ immunotherapy differs between APOE 16. Ashe, K.H. & Zahs, K.R. Neuron 66, 631–645 53. NINDS NET-PD Investigators et al. Neurology 68,
(2010). 20–28 (2007).
ε4 carriers and noncarriers54. Hopefully, 17. Epis, R. et al. Eur. J. Pharmacol. 626, 57–63 (2010). 54. Salloway, S. et al. Neurology 73, 2061–2070
planned efforts to evaluate Parkinson’s disease 18. Kokjohn, T.A. & Roher, A.E. Alzheimers Dement. 5, (2009).
4 advance online publication nature medicine
C O M M E N TA RY
Bridging the Valley of Death of therapeutics
for neurodegeneration
Steven Finkbeiner
Neurodegenerative diseases are the China is predicted to have more citizens with In addition, clinical trials for neurodegen-
sixth leading cause of death in the dementia than the rest of the developed world erative diseases are extraordinarily expensive.
US. The market for disease-modifying combined4. Parkinson’s disease affects 1.0–1.5 The Food and Drug Administration (FDA)
drugs is enormous, but no drug exists. million people in the United States and is also insists that drug trial designs rely heavily on
Academic scientists are increasingly expected to increase two- to threefold by 2050. clinical measures of efficacy. Unfortunately,
pursuing the discovery and development Against this stark backdrop is an even starker clinical measures for neurodegenerative dis-
of therapeutics. Their progress could reality: not a single disease-modifying thera- eases can be relatively insensitive and variable.
potentially reduce the risk of failure peutic exists for the major neurodegenerative Worse still, placebo responses6 measured clini-
sufficiently to warrant greater industry diseases, including Alzheimer’s, Parkinson’s and cally have been increasing over time7, making it
investment and movement of leads Huntington’s diseases and amyotrophic lateral more difficult to detect a beneficial drug effect.
into clinical trials. Here we consider sclerosis (ALS). The need for a drug is urgent. Large numbers of patients must therefore be
the many obstacles to the development The market for a drug for Alzheimer’s and followed for long periods to detect treatment
of therapeutics for neurodegenerative Parkinson’s diseases is enormous and grow- effects. For example, a therapeutic trial for
disease within academia, with a special ing rapidly. Even a drug for orphan diseases, ALS could take 2–3 years because survival is
focus on organizational issues. such as Huntington’s disease or ALS, would be the accepted outcome measure, whereas a trial
highly profitable. Why isn’t there one? And why for rheumatoid or infectious disease might last
is investment in drug development in this area only 3–6 months8.
The imperative so small5? Fortunately, quantitative surrogate measures
It is difficult to discuss the ramifications of The limitations of preclinical models of often detect disease initiation or progression
neurodegenerative diseases for society with- neurodegenerative diseases, the importance more closely than clinical measures. They reveal
out sounding alarmist. In 2010, Alzheimer’s of biomarkers and the difficulties of clinical effects earlier and with fewer patients, thereby
disease, the most common cause of dementia, trials are authoritatively reviewed in this issue. reducing the cost of phase 2 trials to establish
afflicted 5.3 million people and cost $172 bil- Hopefully, the proposed strategies will lead to proof of concept or dosing. Biomarkers custom-
lion in the United States1. Aging is a risk factor breakthroughs. Here I will focus on the orga- designed to detect activity of a specific drug
for Alzheimer’s disease, and the US population nizational obstacles to developing neurothera- candidate on its target can determine whether
over 65 will double by 2030 and quadruple peutics and steps that can be taken today with a putative drug target was engaged during a
by 2050 (ref. 2). Consequently, the number existing tools to reduce the risks (and costs) clinical trial. That way, even a trial that fails
of Americans with Alzheimer’s disease will of drug development for neurodegenerative to demonstrate efficacy will provide valuable
rise to 11–15 million by 2050, unless there is a diseases. information about the disease mechanism.
therapeutic breakthrough1,3. Europe and parts Biomarkers for major neurodegenerative dis-
of Asia face even more daunting challenges Risky business eases are badly needed but are poorly estab-
because, on average, their populations are The risk of drug development is higher for lished and often lacking altogether.
older than those in the United States. By 2040, neurodegeneration than for other diseases. One
reason is that the research and development The Valley of Death
Steven Finkbeiner is at the Gladstone Institute (R&D) tools are mostly unproven. Without Traditionally, industry and academia have
of Neurological Disease, Taube-Koret Center clinically proven therapies, the simpler disease worked together to combat disease. In the
for Huntington’s Disease Research, Consortium models for drug discovery cannot be validated, United States, the National Institutes of Health
for Fronto-temporal Dementia Research, and and the predictive value of any research data (NIH) spends about $28 billion per year to gen-
Departments of Neurology and Physiology, University is unclear. Defining mechanisms of disease is erate biomedical research discoveries. Industry
of California, San Francisco, California, USA. usually a crucial step in identifying drug tar- spends $15–40 billion translating those dis-
e-mail: sfinkbeiner@gladstone.ucsf.edu gets. Drug developers therefore must make big coveries into clinical medicines9. Sometimes
bets on the validity of putative drug targets and industry directly sponsors academic research.
Published online 21 September 2010; perform clinical trials before knowing whether In other cases, industry licenses research tools,
doi: 10.1038/nm.2222 their faith was well placed. technologies or models from academia to
nature medicine advance online publication 1
commentary
facilitate internal drug development programs. try. By contrast, academic career advancement target pharmacologically. Arguably, the indus-
The collaboration has been productive: of the almost universally depends on scholarship, try definition of a ‘druggable’ target may need
21 most important drugs introduced between commonly measured as the frequency and revision, given the decreasing productivity of
1965 and 1992, only five were developed solely impact of publications. Academics jeopardize industry R&D. But as a purely practical mat-
within the private sector10. their careers if they wait to publish a discovery ter, industry remains the major option for a
The last decade has wrought changes in until it is sufficiently developed for industry partnership that would lead to a clinical trial.
this relationship11. Major drug companies are and protected by a patent application. If aca- Industry’s opinion is thus still important.
under tremendous financial pressure. The NIH demic institutions are serious about foster- Even when academics identify and validate
refocused its efforts to encourage academic ing translational research, they need to create a target that industry considers druggable,
researchers to produce more tangible benefits appropriate performance metrics for research- they often lack the capacity to find hits and
for human health12. Increased faculty interest ers, such as invention disclosures, patents and develop them into therapeutic leads. Some
and funding for disease-related research natu- licenses, and perhaps even consider a new institutions have created facilities to perform
rally led to increases in the generation of intel- ‘translational’ faculty track. HTS, but assay options, chemical libraries and
lectual property for academic institutions. The cultural differences are also reflected in medicinal chemistry expertise are limited.
With this confluence of events—drug com- the way research is pursued. Academics are lion- Predictably, the value to industry of leads
panies with funds to invest and the increasing ized for the tenacity, creativity and originality from academic drug discovery programs can
productivity of academic research programs with which they conceive and test hypotheses. be limited. To improve the options for aca-
devoted to finding therapies—why have we In the best case, breakthroughs occur, but often demic researchers, the NIH has created the
failed to see an explosion in new leads going only after focused effort requiring many cycles Molecular Libraries Program (http://mli.nih.
from academia to industry to the clinic? Myriad of grant funding. In the worst case, substantial gov/) and the National Chemical Genomics
obstacles have constipated the flow. The path is academic careers can be spent on misguided Center (NCGC) with a screening capacity
so fraught it has been given a name—the Valley hypotheses that should have been abandoned equivalent to that of a major pharmaceutical
of Death—because many therapeutic strategies long ago. Neither outcome is acceptable in company17. Academic researchers, however,
start the journey but few finish (Fig. 1). industry, where R&D investments must be must still evaluate leads that the NCGC devel-
justified in terms of more immediate returns. ops in secondary assays. Unfortunately, those
What can be done? As a consequence, industry has developed a researchers often lack the capacity to keep the
Despite appearances, the situation is far from culture driven by milestones and punctuated program moving.
hopeless. There are initiatives that can be by performance-based GO/NO-GO decision Manage intellectual property. In 1980, the
pursued today. For example, Congress could points. These cultural differences can create Bayh-Dole Act gave ownership of intellectual
offer sufficiently improved patent protection problems for academics attempting to support property created from US government–funded
to disease-modifying therapies specifically drug discovery efforts in their laboratories with research to the academic institutions where the
for Alzheimer’s and Parkinson’s diseases. To industry-sponsored research agreements15. inventors worked18. Many other countries have
qualify, a provision could be that the new drug The differences in incentives, needs and similar laws. As a result, academic institutions
must produce overall heath care savings13. The resources for researchers in academia and established technology transfer offices (TTOs)
approach would not cost taxpayers anything industry undoubtedly contribute to addi- to decide which inventions to patent, and to
now, and any new drug that qualifies would tional culture gaps. The emphasis on hypoth- formulate terms of licensing agreements.
save taxpayers by lowering the cost of caring esis-driven research and the scarce resources Intellectual property must be protected and
for patients with Alzheimer’s or Parkinson’s available to purse them in academia necessar- licensed under terms that assure industry part-
disease. By changing the risk/benefit ratio to ily lead to small-scale experiments and limited ners of a reasonable return on their substantial
encourage greater investment from the private replicates. Unfortunately, unbiased analyses investment for commercializing an invention.
sector14, we would enable industry to pursue of academic research findings have shown Several common problems afflict TTOs.
earlier-stage leads and thereby build bridges a disappointing degree of reproducibility16. A general lack of industry expertise within
from industry to academia. Industry, on the other hand, must have tightly TTOs often leads to erroneous valuations of
The focus of this article, however, is on ini- reproducible assays to effectively pursue the academic intellectual property19. A positive
tiatives within academia to bridge the Valley of high-throughput screens (HTS) that are the incentive structure to conclude deals is often
Death to industry. It is the thesis here that key lynchpin of early-stage drug discovery. In missing. Fearing deals perceived to be too gen-
components needed to discover and develop academia, an observant scientist can quickly erous to industry, reluctant TTOs may take
effective neurotherapeutics already exist in change course in response to a serendipitous an overly cautious approach that can kill the
academia. Revising and reorganizing these discovery. “In industry,” one expert told me, technology transfer process. The TTO may lack
components could create more effective pro- “serendipity is a failure of quality assurance.” significant flexibility or creativity to structure
grams with the potential to survive the Valley Provide academic researchers with the right agreements to cover patent costs with start-ups
of Death. resources for translational research. Academic or small biotechnology companies, the major-
Reduce the cultural differences between aca- researchers often lack drug discovery resources, ity of licensees15. Unsurprisingly, the licensing
demia and industry. Academia and industry especially expertise, tools and equipment. A rates for intellectual property can be remark-
are separated by a cultural gulf that severely particular molecule or pathway might seem to ably low—in the low single digits at some insti-
limits the ability of academic researchers to be a therapeutic target in a neurodegenerative tutions20.
do translational research. For example, drug disease, but many academics would not know In academia, the inventor frequently grasps
discovery and development are time intensive, whether and how to move these discoveries the significance of the invention and can
but the more intellectual property developed forward. This deficiency can lead to signifi- articulate it much more effectively and with
around a lead program before it becomes pub- cant investment by academics in therapeutic greater credibility than TTO staff. Faculty
lic knowledge, the more valuable it is to indus- strategies that industry knows are difficult to also may have better contacts with potential
2 advance online publication nature medicine
commentary
Drug discovery & development
Traditional purview Traditional purview
Valley of Death
of academia of industry
Assay development Drug development: Preclinical candidate
Target & small molecule hit to lead and (ADME and safety/ First in
identification Target validation screening lead optimization toxicology) human
Traditional NIH basic
tools available to academia
Therapeutics development
research laboratory
Academic and NIH
screening centers
Contract research organizations
Academic non-profit biotechnology incubator
Consortia of
clinicians and researchers
Traditional NIH
& foundation-supported research
Philanthropy and new foundation programs
mechanisms
Funding
New NIH Blueprint programs
Industry-sponsored research and venture capital
Drug development flow
Figure 1 The Valley of Death. Drug discovery and development is an orderly but fraught step-wise process. Traditionally, it has begun with NIH-supported
basic research and the identification of a therapeutic target. Ultimately, it leads to the development of a small molecule whose efficacy is tested in people
by industry. The steps in between are known as the Valley of Death, partly because of the high failure rate and partly because the expertise and resources
available to execute them are critically lacking. Consequently, the vast majority of leads that start the journey fail somewhere along the way. Recently,
individuals and institutions have begun to create new mechanisms to bridge the valley. ADME, absorption, distribution, metabolism and excretion.
partners11,19,21. After licensing, the indus- on the pathogenesis of a particular disease. grants. For example, research programs are
try partner might want the inventor to be Many patient-based foundations also offer often co-directed by a joint research commit-
involved in the development process, and the fellowships and grant support to academics tee of NIH staff and the applicant. Different
time commitment for faculty can be consid- pursuing research into the causes of a particu- notions about drug development from NIH
erable. Participation is strongly influenced by lar neurodegenerative disease. Unfortunately, staff and industry create strains and friction
the extent to which the institution fosters an once these research programs identify potential as the program is executed. Funding rates are
entrepreneurial culture and allocates royal- therapeutic targets, financial support mecha- relatively low, and a lot of time can be spent
ties to the inventor22–25. Still, the career risk is nisms for further development and preclinical fruitlessly on the application process. Some
real for faculty doing substantial translational testing thin out quickly. of the programs require considerable drug-
research, especially those at early stages in their The NIH has recognized this crucial prob- development expertise to construct a compet-
career. lem, especially for orphan diseases. In response, itive application—expertise, as noted earlier,
Focus each partner on what they do best. the National Institute for Neurological Diseases that academics generally lack. Navigating
Translating research discoveries to neurothera- and Stroke has created funding mechanisms to NIH offerings and determining appropriate
peutics involves a lot of moving parts, and the cover every step from basic research and tar- programs can be daunting. The NIH should
mechanisms to fund the process have yet to get identification to first-in-human clinical consider hiring consultants with industry
catch up (Fig. 1). There are three important trials. This development potentially provides expertise to help academics assess the thera-
issues. First, what needs to be done to test the a complete path for academic drug discovery peutic potential of their discoveries, identify
hypothesis underlying the therapeutic strat- and development with federal funds, which do appropriate funding mechanisms to support
egy? Second, can funding be found to reduce not dilute the value of the intellectual prop- development, and construct sensible develop-
risk sufficiently to attract an industry partner erty and therefore do make it more attractive ment programs for neurotherapeutics.
with the resources to develop a discovery into to partners. Patient-based foundations increasingly
a clinical lead? Third, can a license be obtained The NIH still has work to do to realize the want to support drug-discovery programs.
on financial terms that are consistent with the potential of this blueprint for translational They have a unique role in encouraging their
long-term success of a for-profit company? research. Funds are limited: only a few pro- members to participate in clinical studies, and
Ambitious, high-quality academic scien- grams can be supported at once, and those they have had an outsized effect on science by
tists can often attract support from the NIH tend to involve NIH program staff much more using relatively modest fellowship and grant
to conduct a vigorous basic science program intimately than investigator-initiated research support to recruit talented young scientists to
nature medicine advance online publication 3
commentary
Gladstone
Institutes Foundations National
Institutes
of Health
Taube-Koret Center
(Non-profit biotech
incubator) Contract Major
Academia • Biology & chemistry research pharma Therapeutics
• Drug development organizations companies
expertise
• Business development
& technology transfer
Biotechnology
Venture
capital
Figure 2 A model nonprofit biotechnology incubator to facilitate academic drug discovery and development. The Taube-Koret Center acts as a bidirectional
bridge between academia and industry to reduce the risk in the development of therapeutics. Discoveries with therapeutic potential are made in academic
laboratories and are transferred to the Taube-Koret Center within the same institution. The Center maintains the necessary expertise in drug discovery and
medicinal chemistry to validate targets and discover and optimize small molecules to modulate these targets. Contract research organizations are used by
the Center as needed to carry out specific steps in drug development. Leads that fail to meet performance targets are returned to the academic labs that
discovered them for further optimization. Leads that meet milestones are advanced until partners can be found to co-develop them further. The potential
paths to therapeutics via partners (black arrows) can involve foundations, the NIH, existing biotechnology companies, new companies created by venture
capitalists, or major pharmaceutical (pharma) companies. Since the Center’s overarching goal is to find effective therapies for neurodegenerative diseases,
rather than to make a profit from proprietary programs, it can make its new technologies, assays and disease models available to external entities that want to
evaluate the efficacy of their lead programs (gray arrows).
devote their careers to specific diseases. Some and development infrastructure. This unique be a lower-risk mechanism to rapidly advance
have also invested in basic research, without source of support can also be used to make the academic discoveries to a point where they
which many academics could not contemplate academic drug-discovery process much more can, alone or in combination, become the
a rational drug-discovery program. With a few robust and able to sustain programs through basis for an independent entity.
notable exceptions, they generally lack the potentially devastating funding gaps. Similar Regardless of the funding source, trans-
resources and expertise to support and over- to federal support, philanthropy can fuel the parency in constructing research budgets is
see stand-alone drug-discovery programs8. development of therapeutic leads in a way badly needed. The common NIH accounting
One option for foundations is to collaborate that adds substantial value and increases the nomenclature of ‘direct’ and ‘indirect’ costs
and back nonprofit academic drug-discovery potential for partnerships8. has led foundations, donors and VCs to worry
programs at whatever level is feasible. A single Venture capitalists (VCs) and ‘angel inves- that part of their support is used for purposes
foundation might have limited impact, but tors’ have been an important source of finan- unrelated to their interests. Yet institutions
some mechanisms of neurodegeneration, for cial support for commercializing academic cannot commit to do work that incurs finan-
example, might be common. Collaboration research28. VCs supply substantial capital, cial loss. One solution is to change the struc-
among foundations might enable their par- business development expertise, and indus- ture of research budgets so that they include
ticipation in serious drug discovery efforts try contacts that catalyze the development of all project-related costs and provide enough
and lead to synergies that help everyone and an academic discovery at a pace that would detail so that donors can judge for themselves
leverage their investment. otherwise be impossible. Unfortunately, the whether their support is being used properly
In this context, major philanthropists might recent global financial crisis and the reduced and is tailored to the project.
have the most important role in enabling drug opportunities for initial public offerings Academic institutions must be transpar-
discovery in academic settings. Generally, they have affected VC investment in biotech- ent and able to deliver on programs they
are motivated because they or a family mem- nology. Today, VCs are making fewer new create. Neurodegenerative diseases unfold
ber have been affected by or are at risk for investments, and they tend to target discov- slowly. Consequently, the time that is neces-
a particular disease. The total cost of devel- eries at a later stage of development. Large sarily required to perform experiments and
oping a new drug in 2000 was estimated to pharmaceutical companies have become the make iterative progress with animal mod-
be $800 million26,27, but only one-third of main customers for VCs, which also affects els of these slow diseases can be frustrating
the estimated early-stage costs of preclinical the disease indications that VCs pursue. VCs for philanthropists and foundations with-
development were direct costs. The remaining should explore new, less risky investment out backgrounds in biology, who need to
two-thirds were ‘opportunity costs’ of tying models. For example, rather than founding be assured by external review mechanisms
up capital for an extended drug-development new companies with expensive facilities and that their investment is a good one. More
campaign. Used strategically, philanthropic management structures, VCs should explore important, academic institutions must see
support can be leveraged five- to tenfold smaller, targeted investments to back devel- the drug-discovery effort as part of their
to attract support from the NIH or private opment within the inventor’s laboratory. The mission. Donations that support this pro-
sources to create a substantial drug-discovery creation of commercial incubators might also cess may then result in unrestricted revenue
4 advance online publication nature medicine
commentary
for the academic institution from the extra chemistry capabilities on site, but these capa- cial development opportunity. Instead, the
licenses that are generated. bilities focus on in silico screening and analysis, incubator can transfer the program back to
on complex, small-scale small-molecule syn- the academic laboratory from which it came
Two experimental models theses, and on addressing the bioavailability for further optimization.
Academia is working to implement these challenge posed by the blood-brain barrier. Notably, the goal of the incubator is to com-
ideas and to create innovative structures with Simpler analog chemistry or scale-up chemis- plement, not replace, biotechnology or phar-
the capability to reduce the risk of developing try is done through CROs with guidance from maceutical company activities by generating
neurotherapeutics and successfully navigate our consulting medicinal chemists. CROs are promising leads and sufficiently ‘de-risking’
the Valley of Death. Two examples are offered also used for absorption, distribution, metab- them to warrant partnerships. As such, the
below. olism, excretion, toxicology and other studies incubator is freer to pursue innovative thera-
The Taube-Koret Center: a nonprofit required for later-stage preclinical drug devel- peutic targets for Alzheimer’s or Parkinson’s
biotechnology incubator. At the Gladstone opment. disease that may be seen as too risky for early-
Institutes, we established a center that func- As a nonprofit incubator, TKC’s overarch- stage R&D by industry or to pursue disease
tions as a nonprofit biotechnology incubator ing goal is to find therapeutics for neurode- indications, such as Huntington’s disease or
to develop therapeutics for neurodegenerative generative disease. This has important policy ALS, that are sometimes seen as too small to
disease (Fig. 2). It offers a model of how drug implications. First, TKC is purposely struc- attract early-stage VC investment. If success-
discovery can be conducted effectively in an tured to identify and investigate as many ful, both goals should lead to more and bet-
academic setting and suggests collaborating therapeutic targets and leads as possible in ter therapeutic options for neurodegenerative
roles for the institution and philanthropists. the belief that, as drug discovery is inherently disease.
The Taube-Koret Center (TKC; http://taube- risky, the more leads that can be developed, Alzheimer’s Disease Neuroimaging
koret-center.org/) was established with phil- the better the chance of finding one that works Initiative: a model public-private partnership
anthropic support from the Koret Foundation in a reasonable time. Partnerships bring cru- focused on biomarkers. Lack of good biomark-
and Taube Philanthropies, with an initial focus cial expertise and free up internal resources. ers contributes to the high cost of clinical tri-
on Huntington’s disease. Several therapeutic Therefore, leads are partnered at the earliest als, which limits the number of studies and
leads from this program have shown promise stage possible. Should a partnership fail after the companies pursuing neurodegenerative
in models of other diseases, such as Alzheimer’s it is initiated, the rights to develop the lead disease overall. The investment needed to find
disease, Parkinson’s disease, ALS and fronto- revert to TKC. Because our goal is to find a biomarkers can be so high that, for the NIH
temporal dementia, so other donors are joining therapeutic, we do not restrict access to the to take it on alone, support for other research
to expand its scope. TKC to homegrown leads. The TKC has estab- would be severely reduced. Similarly, com-
Although the TKC operates similarly to a lished models of neurodegeneration based on panies have difficulty justifying biomarker
biotechnology company, it resides within a mutant huntingtin, TDP-43, SOD1, LRRK2, programs, given the uncertain and long-term
nonprofit research institute. Potential neuro- synuclein and APP and has invented platform nature of the potential benefit.
therapeutic leads mostly come from NIH- and technology to conduct drug discovery in pri- In 2004, the Alzheimer’s Disease
foundation-supported basic research programs mary cells. These resources have been made Neuroimaging Initiative (ADNI) was formed
of two investigators at the same institution. The available to biotechnology or major pharma- to investigate relationships among clinical,
center is physically separate from the main aca- ceutical companies who have small-molecule cognitive, imaging, genetic and biochemical
demic labs, allowing a somewhat distinct, more leads to test. biomarkers characteristic of individuals devel-
industry-like environment. It also provides aca- What advantages for CNS drug discovery oping Alzheimer’s disease29,30. These data will
demics an opportunity to contribute discoveries might a nonprofit academic biotechnology help clinicians to diagnose Alzheimer’s disease,
to the Center for further development without incubator have over a for-profit biotechnol- validate biomarkers for disease progression and
obliging them to do drug discovery themselves, ogy company? First, an academic incubator design better clinical trials30. This extremely
if their interests and strengths lie elsewhere. A is shielded from the structure imposed on ambitious study has received $60 million: $40
consultant experienced in drug-development for-profit companies by investors, which is million from the National Institutes on Aging
reviews potential targets from these research geared to provide a timely financial return. and $20 million from 13 pharmaceutical com-
programs and advises on milestones and per- The financial depth and stability of major aca- panies and two foundations. The study now
formance-based GO/NO-GO decision points. demic institutions represent a more enduring involves academic researchers at 58 sites in the
Leads that fail to meet performance milestones structure than many biotechnology companies. United States and Canada.
are replaced with the next most promising As a result, pursuit of optimal drug discovery ADNI is impressive for many reasons, not
leads. The consultant, who has the necessary can drive decisions, and philanthropists and least of which is that it was conducted by a
experience to negotiate agreements in the area foundations can invest with confidence. The consortium of highly successful researchers29.
of neurodegeneration research, works closely incubator avoids incurring early-stage trans- Remarkably, to accelerate research, the aca-
with the TTO to develop an overall intellectual actional costs to acquire intellectual property demics leading the cooperative project agreed
property strategy with greater value for poten- normally borne by biotechnology companies to make their unpublished data publicly avail-
tial industry partners by protecting later-stage because the feeder academic laboratories are able soon after it is collected. Research teams
intellectual property. at the same institution. These laboratories also that are not formally part of ADNI have even
TKC conducts drug discovery and devel- provide the incubator with a diverse ‘pipeline’, analyzed and published using ADNI data.
opment through a combination of internal ensuring that efforts can be sustained even as Historically, building research consortia has
programs and external contract research orga- specific lead programs fail. Constrained by been challenging. Academic career advance-
nizations (CROs). For example, target identi- conventional GO/NO-GO decision points, ment is substantially based on contributions
fication, validation, HTS and in vivo efficacy biotechnology companies might abandon of individuals to their chosen scientific field.
trials are all done on site. TKC has medicinal leads prematurely and miss a potentially cru- Assessing those contributions within large con-
nature medicine advance online publication 5
commentary
sortia is difficult, and promotion committees will face back-breaking obligations to Medicare 270 (2009).
4. Ferri, C.P. et al. Lancet 366, 2112–2117 (2005).
struggle to assign values to multiauthored con- and Medicaid unless a cost-saving treatment is 5. Acemoglu, D. & Linn, J. Q. J. Econ. 119, 1049–1090
sortium publications. Indeed, some institutions found32. More funding is critically needed for (2004).
instill an anticollaborative culture, and team the National Institute for Neurological Diseases 6. Benedetti, F., Carlino, E. & Pollo, A.
Neuropsychopharmacology published online,
science is viewed negatively. Unfortunately, and Stroke and the National Institute on Aging, doi:10.1038/npp.2010.81 (30 June 2010).
some problems in neurodegeneration are so the NIH institutes primarily responsible for 7. Spiegel, A. The growing power of the sugar pill. National
Public Radio <http://www.npr.org/templates/story/story.
large it is hard to imagine solving them with- basic and translational research in neuro- php?storyId=124367058> (2010).
out large teams of collaborating scientists. It is degenerative diseases. The NIH has moved 8. Hanson, S., Nadig, L. & Altevogt, B. Venture philan-
encouraging to see that the ADNI approach has resources into supporting drug discovery and thropy strategies to support translational research:
workshop summary <http://www.nap.edu/cata-
been replicated in other countries. Hopefully, development within academia; yet, opportu- log/12558.html> (Forum on Neuroscience and Nervous
cultural changes are under way within aca- nities vastly exceed resources. VCs have been System Disorders, Institute of Medicine of the National
Academies, 2009).
demic institutions to place a higher value on leery of early-stage drug discovery for neuro- 9. Congressional Budget Office. Research and develop-
team science. degenerative disease, but they have recently ment in the pharmaceutical industry (US Government
Recognizing that the establishment of bio- been exploring new and more cost-effective Printing Office, 2006).
10. Cockburn, I.M. & Henderson, R.M. in NBER Innovation
markers is risky but necessary for developing ways to fund early-stage development efforts. Policy and the Economy vol. 1 (eds. Jaffe, A.B., Lerner,
therapeutics for Alzheimer’s disease, ADNI Philanthropists may have the greatest opportu- J. & Stern, S.) 20–21 (National Bureau of Economic
Research, Cambridge, Massachusetts, USA, 2000).
has spread the risks and benefits among many nity and ability to materially enable academic 11. Young, T.A. Int. J. Intellect. Prop. Law Econ. Manage.
interested partners. More broadly, the approach drug discovery and development. 1, 13–18 (2005).
highlights the idea of identifying precompeti- Neurodegenerative diseases are not going 12. National Institutes of Health. Blueprint neurotherapeu-
tics network. NIH Blueprint for Neuroscience Research
tive space in neurodegeneration research that away. The cost of ignoring them will break our <http://neuroscienceblueprint.nih.gov/bpdrugs/index.
could be successfully pursued by international fragile healthcare systems. Our only hope is htm> accessed 23 August 2010.
13. Duggan, M. J. Health Econ. 24, 1–31 (2005).
consortia and funded with pooled resources through focused research to find therapies. With 14. Lanjouw, J.O. & Cockburn, I.M. World Dev. 29, 265–
from public and private sources8. Perhaps the concerted action from academia, the govern- 289 (2001).
recently announced agreement among major ment and philanthropy, the risks in developing 15. Fraser, J. Tomm. Technol. Trans. 1, 9–20 (2009).
16. Ioannidis, J.P.A. PLoS Clin. Trials 2006, e36 (2006).
pharmaceutical companies to share data from those therapies can be substantially lowered. In 17. Inglese, J. et al. Proc. Natl. Acad. Sci. USA 103,
11 failed clinical trials in Alzheimer’s disease turn, industry will respond with greater atten- 11473–11478 (2006).
18. Boettiger, S. & Bennett, A.B. Nat. Biotechnol. 24,
will also provide valuable insights that will lead tion and investment, and therapies will result.
320–323 (2006).
to more successful future clinical trials, even 19. Thursby, J. & Thursby, M. AUTM J. 12, 9–22 <http://
ACKNOWLEDGMENTS www.provendis.info/fileadmin/info/pdfs/1255.pdf>
if this initiative is more an act of desperation
This work was supported by the Taube-Koret (2000).
than a new dawn of genuine collaboration31. Center for Huntington’s Disease Research, by NIH - 20. Thursby, J., Jensen, R. & Thursby, M. J. Technol.
National Institute of Neurological Disorders and Transf. 26, 59–72 (2001).
Summary Stroke grants 2R01 NS039074 and 2R01 NS045091 21. Lockett, A. & Wright, M. Res. Policy 34, 1043–1057
and NIH - National Institute on Aging grant 2P01 (2005).
Developing therapies for neurodegenera- 22. Friedman, J. & Silberman, J. J. Technol. Transf. 28,
AG022074, and by The J. David Gladstone Institutes.
tive diseases is extremely challenging. As the 17–30 (2003).
I thank S. Williams, S. Freedman, M. Sutherland, 23. Markman, G., Phan, P., Balkin, D. & Gianiodis, P.
main developer of therapies, the for-profit F. Dorey, M. Lopez, L. Bruijn, R. Blumenstein, R. J. Technol. Transf. 29, 353–364 (2004).
pharmaceutical industry has done the calcula- Pacifici, N. Wexler, C. Johnson, C. Austin, L. Vetter, 24. Lach, S. & Schankerman, M. J. Euro. Econ. Assoc. 2,
tion. Although a therapy for neurodegenera- A. Grove, P. Lansbury, T. Reisine, G. Naeve and 252–264 (2004).
members of the Finkbeiner laboratory for helpful 25. Link, A.N. & Siegel, D.S. Eur. J. Finance 11, 169–181
tive disease would be highly profitable, it has (2005).
discussions and/or comments on an early version of
determined that its R&D dollar can be spent 26. DiMasi, J.A., Hansen, R.W. & Grabowski, H.G.
this manuscript. I thank G. Howard and S. Ordway for
J. Health Econ. 22, 151–185 (2003).
on lead programs for other indications that are editorial assistance, and K. Nelson for administrative 27. Adams, C.P. & Brantner, V.B. Health Aff. 25, 420–428
less risky. assistance. (2006).
In view of the looming demographic 28. Reinhardt, U.E. Health Aff. 20, 136–149 (2001).
COMPETING FINANCIAL INTERESTS 29. Aisen, P.S. et al. Alzheimers Dement. 6, 239–246
changes, action on neurodegenerative diseases The authors declare no competing financial interests. (2010).
is urgent. Every player has a role. Academic 30. Weiner, M.W. et al. Alzheimers Dement. 6, 202–211
1. Alzheimer’s Association. Alzheimers Dement. 6, 158– (2010).
institutions need to demonstrate their com- 31. Anonymous. Economist 395, 81–82 (17 June
194 (2010).
mitment to translational research by changing 2. Centers for Disease Control and Prevention and The 2010).
the cultural impediments that force academic Merck Company Foundation. The state of health 32. Bynum, J. Characteristics, costs, and health service
and aging in America (Merck Company Foundation, use for Medicare beneficiaries with a dementia diag-
researchers to abandon these lines of investiga- Whitehouse Station, New Jersey, USA, 2007). nosis. Report 1: Medicare current beneficiary survey
tion or leave academia for industry. Congress 3. Alzheimer’s Association. Alzheimers Dement. 5, 234– (Alzheimer’s Association, 2009).
6 advance online publication nature medicine
Das könnte Ihnen auch gefallen
- Seminar: Kaj Blennow, Mony J de Leon, Henrik ZetterbergDokument17 SeitenSeminar: Kaj Blennow, Mony J de Leon, Henrik ZetterbergDanni XNoch keine Bewertungen
- Klein 2006Dokument13 SeitenKlein 2006Dani AlmeidaNoch keine Bewertungen
- Murphy 2010Dokument13 SeitenMurphy 2010Nadia SaiNoch keine Bewertungen
- Stem Cell Treatment Alzheimer's Disease - ASCI - Asian Stem Cell InstituteDokument39 SeitenStem Cell Treatment Alzheimer's Disease - ASCI - Asian Stem Cell Institutea73072Noch keine Bewertungen
- Cross interactions between the Alzheimer's disease amyloid-β peptide and other amyloid proteins: a further aspect of the amyloid cascade hypothesisDokument17 SeitenCross interactions between the Alzheimer's disease amyloid-β peptide and other amyloid proteins: a further aspect of the amyloid cascade hypothesisMAYRA PAZNoch keine Bewertungen
- Perspective Is Therapeutic Plasma Exchange A Viable Option For TreatingDokument11 SeitenPerspective Is Therapeutic Plasma Exchange A Viable Option For TreatingRaul Rios RitterNoch keine Bewertungen
- β-amyloid Peptides and Amyloid Plaques in Alzheimer's DiseaseDokument9 Seitenβ-amyloid Peptides and Amyloid Plaques in Alzheimer's DiseaseNarcisa MateiNoch keine Bewertungen
- Biol 317 Term PaperDokument9 SeitenBiol 317 Term PaperSohret PektuncNoch keine Bewertungen
- Ciclo Celular y CáncerDokument12 SeitenCiclo Celular y CáncerAlfonso De la FuenteNoch keine Bewertungen
- Alpha-Synuclein Secretion and DysfunctionDokument7 SeitenAlpha-Synuclein Secretion and Dysfunctionbenghoe77Noch keine Bewertungen
- THE GENETICS OF ALZHEIMERS DISEASE. PPTDokument24 SeitenTHE GENETICS OF ALZHEIMERS DISEASE. PPTBenjaminVasileniucNoch keine Bewertungen
- Alves Et Al - 2016 - Gene Therapy Strategies For Alzheimer's DiseaseDokument8 SeitenAlves Et Al - 2016 - Gene Therapy Strategies For Alzheimer's DiseaseMaria ChenNoch keine Bewertungen
- Review Nature AD OrganoidiDokument15 SeitenReview Nature AD Organoidifabiofab4Noch keine Bewertungen
- Neurochemical Aspects of Alzheimer's Disease: Risk Factors, Pathogenesis, Biomarkers, and Potential Treatment StrategiesVon EverandNeurochemical Aspects of Alzheimer's Disease: Risk Factors, Pathogenesis, Biomarkers, and Potential Treatment StrategiesBewertung: 3.5 von 5 Sternen3.5/5 (3)
- Leading Edge Review: Alzheimer Mechanisms and Therapeutic StrategiesDokument19 SeitenLeading Edge Review: Alzheimer Mechanisms and Therapeutic StrategiesArtemis LiNoch keine Bewertungen
- Genes, Environment and Alzheimer's DiseaseVon EverandGenes, Environment and Alzheimer's DiseaseOrly LazarovNoch keine Bewertungen
- European Journal of Internal Medicine: Stephanie Seneff, Glyn Wainwright, Luca MascitelliDokument7 SeitenEuropean Journal of Internal Medicine: Stephanie Seneff, Glyn Wainwright, Luca MascitelliApril ayuNoch keine Bewertungen
- Therapeutic Plasma Exchange With Albumin: A New Approach To Treat Alzheimer's DiseaseDokument8 SeitenTherapeutic Plasma Exchange With Albumin: A New Approach To Treat Alzheimer's DiseaseAli Umri HasibuanNoch keine Bewertungen
- VPS35 Haploinsufficiency - AD Early OnsetDokument15 SeitenVPS35 Haploinsufficiency - AD Early OnsetChunlei WangNoch keine Bewertungen
- Multiple System Atrophy: Cellular and Molecular Pathology: D J Burn, E JarosDokument8 SeitenMultiple System Atrophy: Cellular and Molecular Pathology: D J Burn, E Jarosuzair khanNoch keine Bewertungen
- Immunity & Ageing: Alzheimer's Disease: New Diagnostic and Therapeutic ToolsDokument5 SeitenImmunity & Ageing: Alzheimer's Disease: New Diagnostic and Therapeutic ToolsmarcussiNoch keine Bewertungen
- Yoshiyama 2012Dokument13 SeitenYoshiyama 2012yusufNoch keine Bewertungen
- Msa NejmDokument15 SeitenMsa NejmSantosh DashNoch keine Bewertungen
- Multiple-System AtrophyDokument15 SeitenMultiple-System AtrophyNathasia SuryawijayaNoch keine Bewertungen
- Alzheimer Disease EngDokument35 SeitenAlzheimer Disease EngAna IantucNoch keine Bewertungen
- A New Understanding of Alzheimer's (Scientific American, Aug 2021)Dokument5 SeitenA New Understanding of Alzheimer's (Scientific American, Aug 2021)No Spam MeNoch keine Bewertungen
- MiscellaneousDokument4 SeitenMiscellaneouszeeshanNoch keine Bewertungen
- Compendium of Selected Recent Publications Cell and Molecular Biology ResearchDokument16 SeitenCompendium of Selected Recent Publications Cell and Molecular Biology ResearchXyza Kim OlivaNoch keine Bewertungen
- Alzheimer DiseaseDokument16 SeitenAlzheimer DiseaseHabib G. Moutran BarrosoNoch keine Bewertungen
- Neurodegeneration: The Molecular Pathology of Dementia and Movement DisordersVon EverandNeurodegeneration: The Molecular Pathology of Dementia and Movement DisordersDennis DicksonNoch keine Bewertungen
- AD Review 2Dokument14 SeitenAD Review 2Emmanuel JacquezNoch keine Bewertungen
- AlzheimersDokument127 SeitenAlzheimersshanes100% (1)
- Aurt2011 398636Dokument16 SeitenAurt2011 398636Ilana FuxNoch keine Bewertungen
- Alzheimer PatohDokument27 SeitenAlzheimer PatohHenry Leroy Lewis BatresNoch keine Bewertungen
- NIH Public Access: Abeta, Oxidative Stress in Alzheimer Disease: Evidence Based On Proteomics StudiesDokument27 SeitenNIH Public Access: Abeta, Oxidative Stress in Alzheimer Disease: Evidence Based On Proteomics StudiesNarcisa MateiNoch keine Bewertungen
- Neuroinflammation, Gut Microbiome, and Alzheimer 'S DiseaseDokument8 SeitenNeuroinflammation, Gut Microbiome, and Alzheimer 'S DiseaseChristina MountakiNoch keine Bewertungen
- Discussing The Role of Tau & Amyloid in Alzheimer's (2018)Dokument18 SeitenDiscussing The Role of Tau & Amyloid in Alzheimer's (2018)Brittany JordanNoch keine Bewertungen
- Neural ApoptosisDokument6 SeitenNeural ApoptosisJay HardkickNoch keine Bewertungen
- Alzheimers Disease Research Paper ConclusionDokument7 SeitenAlzheimers Disease Research Paper Conclusiongtnntxwgf100% (1)
- Late-Onset Alzheimers Disease Is Associated WithDokument13 SeitenLate-Onset Alzheimers Disease Is Associated WithAlix AliNoch keine Bewertungen
- Alzheimers Biology CourseworkDokument8 SeitenAlzheimers Biology Courseworkpqltufajd100% (2)
- Amyloid-β in Alzheimer's Disease: Effects on Disease Pathogenesis and Recent Advances in Clinical TrialsDokument8 SeitenAmyloid-β in Alzheimer's Disease: Effects on Disease Pathogenesis and Recent Advances in Clinical TrialsInternational Journal of Innovative Science and Research TechnologyNoch keine Bewertungen
- Amyotrophic Lateral Sclerosis - Mechanisms and Therapeutics in The Epigenomic EraDokument14 SeitenAmyotrophic Lateral Sclerosis - Mechanisms and Therapeutics in The Epigenomic EraEduardo Martinez VillalvazoNoch keine Bewertungen
- Artículo 2Dokument6 SeitenArtículo 2Gabby Hernandez RodriguezNoch keine Bewertungen
- Neuroimmunology: Multiple Sclerosis, Autoimmune Neurology and Related DiseasesVon EverandNeuroimmunology: Multiple Sclerosis, Autoimmune Neurology and Related DiseasesAmanda L. PiquetNoch keine Bewertungen
- 1 s2.0 S0925443911002754 Main PDFDokument7 Seiten1 s2.0 S0925443911002754 Main PDFMaria SousaNoch keine Bewertungen
- Protein Misfolding and NeurodegenerationDokument6 SeitenProtein Misfolding and NeurodegenerationDaniel RomeroNoch keine Bewertungen
- Alzheimer's - Alzheimer's Disease - Kaj Blennow, Mony J de Leon, Henrik ZeterbergDokument17 SeitenAlzheimer's - Alzheimer's Disease - Kaj Blennow, Mony J de Leon, Henrik ZeterbergCatinean DanielaNoch keine Bewertungen
- What Is It Behind Alzheimer's Disease? A Pathophysiology ReviewDokument8 SeitenWhat Is It Behind Alzheimer's Disease? A Pathophysiology ReviewAnonymous 4iaWWhhDU8Noch keine Bewertungen
- The Amyloid Hypothesis of Alzheimer's Disease at 25 YearsDokument14 SeitenThe Amyloid Hypothesis of Alzheimer's Disease at 25 YearsNguyễn Thị Thảo NhungNoch keine Bewertungen
- Amyloid - Induced Neuronal Dysfunction in Alzheimer's Disease: From Synapses Toward Neural NetworksDokument7 SeitenAmyloid - Induced Neuronal Dysfunction in Alzheimer's Disease: From Synapses Toward Neural NetworkskleberkillerNoch keine Bewertungen
- Department of Physiology Master of Science in PhysiologyDokument77 SeitenDepartment of Physiology Master of Science in Physiologyrichardmd2100% (1)
- Biologia Da Mielina e LeucodistrofiasDokument16 SeitenBiologia Da Mielina e LeucodistrofiasFernando Cezar dos SantosNoch keine Bewertungen
- Alzheimer'S Disease: Preseented by Arun Varughese GroupDokument33 SeitenAlzheimer'S Disease: Preseented by Arun Varughese GroupManisanthosh KumarNoch keine Bewertungen
- Molecules: - Amyloid and The Pathomechanisms of Alzheimer's Disease: A Comprehensive ViewDokument32 SeitenMolecules: - Amyloid and The Pathomechanisms of Alzheimer's Disease: A Comprehensive ViewdianawilcoxNoch keine Bewertungen
- Effects of Neural Stem Cell TransplantationDokument11 SeitenEffects of Neural Stem Cell TransplantationChouaib OujhainNoch keine Bewertungen
- Rapid Review: BackgroundDokument9 SeitenRapid Review: BackgroundyusufNoch keine Bewertungen
- Sorting Receptor SORLA - A Trafficking Path To Avoid Alzheimer DiseaseDokument10 SeitenSorting Receptor SORLA - A Trafficking Path To Avoid Alzheimer DiseaseC MNoch keine Bewertungen
- 111Dokument1 Seite111schizoid.paranoidNoch keine Bewertungen
- 666Dokument1 Seite666schizoid.paranoidNoch keine Bewertungen
- 111Dokument1 Seite111schizoid.paranoidNoch keine Bewertungen
- 111Dokument1 Seite111schizoid.paranoidNoch keine Bewertungen
- Novo Documento de TextoDokument1 SeiteNovo Documento de Textoschizoid.paranoidNoch keine Bewertungen
- 111Dokument1 Seite111schizoid.paranoidNoch keine Bewertungen
- 111Dokument1 Seite111schizoid.paranoidNoch keine Bewertungen
- 111Dokument1 Seite111schizoid.paranoidNoch keine Bewertungen
- 111Dokument1 Seite111schizoid.paranoidNoch keine Bewertungen
- 111Dokument1 Seite111schizoid.paranoidNoch keine Bewertungen
- 111Dokument1 Seite111schizoid.paranoidNoch keine Bewertungen
- Novo Documento de TextoDokument1 SeiteNovo Documento de Textoschizoid.paranoidNoch keine Bewertungen
- 111Dokument1 Seite111schizoid.paranoidNoch keine Bewertungen
- 111Dokument1 Seite111schizoid.paranoidNoch keine Bewertungen
- 111Dokument1 Seite111schizoid.paranoidNoch keine Bewertungen
- 111Dokument1 Seite111schizoid.paranoidNoch keine Bewertungen
- Pub Med ArticleDokument2 SeitenPub Med Articleschizoid.paranoidNoch keine Bewertungen
- Novo Documento de TextoDokument1 SeiteNovo Documento de Textoschizoid.paranoidNoch keine Bewertungen
- Novo Documento de TextoDokument1 SeiteNovo Documento de Textoschizoid.paranoidNoch keine Bewertungen
- Hitler PDF UnproofDokument16 SeitenHitler PDF UnproofYusup RamdaniNoch keine Bewertungen
- Novo Documento de TextoDokument1 SeiteNovo Documento de Textoschizoid.paranoidNoch keine Bewertungen
- Novo Documento de TextoDokument1 SeiteNovo Documento de Textoschizoid.paranoidNoch keine Bewertungen
- Personality Disorders TCCDokument3 SeitenPersonality Disorders TCCschizoid.paranoidNoch keine Bewertungen
- Psychopathy and Antisocial Personality Disorder A Case of Diagnostic ConfusionDokument7 SeitenPsychopathy and Antisocial Personality Disorder A Case of Diagnostic Confusionschizoid.paranoid100% (1)
- Change LogDokument2 SeitenChange LogKriszorNoch keine Bewertungen
- An fMRI Study of Facial Emotion Processing in Patients With SchizophreniaDokument8 SeitenAn fMRI Study of Facial Emotion Processing in Patients With Schizophreniaschizoid.paranoidNoch keine Bewertungen
- NIH Public Access: Author ManuscriptDokument70 SeitenNIH Public Access: Author Manuscriptschizoid.paranoidNoch keine Bewertungen
- Autistic Disorders and Schizophrenia Related or Remote An Anatomical Likelihood EstimationDokument8 SeitenAutistic Disorders and Schizophrenia Related or Remote An Anatomical Likelihood Estimationschizoid.paranoidNoch keine Bewertungen
- Mqs For Mrcog Part 2Dokument174 SeitenMqs For Mrcog Part 2Poppy HazarikaNoch keine Bewertungen
- CTE StudyDokument7 SeitenCTE StudyTom LeyNoch keine Bewertungen
- Materi Young Onset Demensia (YOD) DR - YudaDokument20 SeitenMateri Young Onset Demensia (YOD) DR - Yudacimoy kwNoch keine Bewertungen
- Carol Turkington Joseph R Harris The Encyclopedia of Memory and Memory Disorders Facts On File Library of Health and Living Facts On File 2001Dokument305 SeitenCarol Turkington Joseph R Harris The Encyclopedia of Memory and Memory Disorders Facts On File Library of Health and Living Facts On File 2001AmalNoch keine Bewertungen
- 7996 PDFDokument24 Seiten7996 PDFChristopher WellsNoch keine Bewertungen
- Manual Psicoger SparDokument430 SeitenManual Psicoger Sparpereiraboan9140Noch keine Bewertungen
- Effects of Neural Stem Cell TransplantationDokument11 SeitenEffects of Neural Stem Cell TransplantationChouaib OujhainNoch keine Bewertungen
- Natural Treatments For Alzheimers 35ppDokument35 SeitenNatural Treatments For Alzheimers 35ppaman_arora100% (1)
- PN0621 CF1 ClinicalApproachDementiaDokument5 SeitenPN0621 CF1 ClinicalApproachDementiaSwayang Sudha PandaNoch keine Bewertungen
- EXERCISE Last DecDokument9 SeitenEXERCISE Last DecAzahwa 84Noch keine Bewertungen
- Neurotoxicology: Mona Hersi, Brittany Irvine, Pallavi Gupta, James Gomes, Nicholas Birkett, Daniel KrewskiDokument45 SeitenNeurotoxicology: Mona Hersi, Brittany Irvine, Pallavi Gupta, James Gomes, Nicholas Birkett, Daniel KrewskiMAYRA PAZNoch keine Bewertungen
- Worry More Live Longer British English Teacher Ver2Dokument7 SeitenWorry More Live Longer British English Teacher Ver2aisthesicNoch keine Bewertungen
- COVID-19 Information: CDC, NIH, NCBI, HHS ResourcesDokument2 SeitenCOVID-19 Information: CDC, NIH, NCBI, HHS ResourcesNaïs LABBADINoch keine Bewertungen
- Improve Your Brain Health: HealthmeansDokument34 SeitenImprove Your Brain Health: HealthmeansAnnica MediciNoch keine Bewertungen
- Rupal Arora - Diagnostic Criteria For Neuropychological DisordersDokument21 SeitenRupal Arora - Diagnostic Criteria For Neuropychological Disordersrupal aroraNoch keine Bewertungen
- Letters Combined (11 Referral, 2 Discharge, 1 Transfer)Dokument49 SeitenLetters Combined (11 Referral, 2 Discharge, 1 Transfer)Faisal ImtiazNoch keine Bewertungen
- (Đề thi gồm 04 trang) Thời gian làm bài. 60 phút, không kể thời gian phát đềDokument4 Seiten(Đề thi gồm 04 trang) Thời gian làm bài. 60 phút, không kể thời gian phát đềdml_qtriNoch keine Bewertungen
- Can Marijuana Help With or Prevent Alzheimer'sDokument10 SeitenCan Marijuana Help With or Prevent Alzheimer'sLorina BoligNoch keine Bewertungen
- An Arabic Version of The Cognitive Subscale of The Alzheimer's Disease Assessment Scale (ADAS-Cog) - Reliability, Validity and Normative DateDokument11 SeitenAn Arabic Version of The Cognitive Subscale of The Alzheimer's Disease Assessment Scale (ADAS-Cog) - Reliability, Validity and Normative DateTarekSadokNoch keine Bewertungen
- How To Remember Things - 21 Proven Memory Techniques PDFDokument53 SeitenHow To Remember Things - 21 Proven Memory Techniques PDFFawad AhmadNoch keine Bewertungen
- Complete Alzheimer S CureDokument7 SeitenComplete Alzheimer S CureJ100% (1)
- NCP IDNT Terminology ChangesDokument59 SeitenNCP IDNT Terminology ChangesFerdian Hapreda JanuardoNoch keine Bewertungen
- Functional Cognitive Disorder: Dementia's Blind Spot: UpdateDokument9 SeitenFunctional Cognitive Disorder: Dementia's Blind Spot: UpdatePriscila Lorencini SelingardiNoch keine Bewertungen
- Scientific American - October 2014 USADokument104 SeitenScientific American - October 2014 USAmarproofNoch keine Bewertungen
- BADAC FlyerDokument1 SeiteBADAC FlyerAlbert JoseNoch keine Bewertungen
- Alzheimer's - Alzheimer's Disease - Kaj Blennow, Mony J de Leon, Henrik ZeterbergDokument17 SeitenAlzheimer's - Alzheimer's Disease - Kaj Blennow, Mony J de Leon, Henrik ZeterbergCatinean DanielaNoch keine Bewertungen
- Research criteria for Alzheimer's disease diagnosisDokument13 SeitenResearch criteria for Alzheimer's disease diagnosisEuge López RuizNoch keine Bewertungen
- Personality Changes in Dementia: by Sonya Laputz Care SpecialistDokument2 SeitenPersonality Changes in Dementia: by Sonya Laputz Care Specialistlove punjabNoch keine Bewertungen
- Dementia: I Wayan Tunjung, DR - Sp.S. Bagian Neurologi RSU Kota MataramDokument67 SeitenDementia: I Wayan Tunjung, DR - Sp.S. Bagian Neurologi RSU Kota MataramHerlinawatiHariniNoch keine Bewertungen
- Board Review Geriatrics: Manage Depression with Increased Sertraline DoseDokument95 SeitenBoard Review Geriatrics: Manage Depression with Increased Sertraline DoseCharis Paroginog100% (2)
- Aging Quiz 2 AnswersDokument5 SeitenAging Quiz 2 AnswersdindandinigaNoch keine Bewertungen