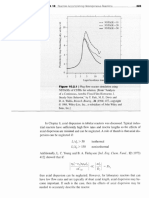
Das könnte Ihnen auch gefallen
- The Toxicology of Radioactive Substances: Volume 5Von EverandThe Toxicology of Radioactive Substances: Volume 5A. A. LetavetNoch keine Bewertungen
- Materials and MethodsDokument6 SeitenMaterials and Methodsفقوري البصراويNoch keine Bewertungen
- Hotz 1971Dokument5 SeitenHotz 1971CoNoch keine Bewertungen
- 103355.1 20201218131355 CoveredDokument11 Seiten103355.1 20201218131355 CoveredBhavani SenthilkumarNoch keine Bewertungen
- Potassium: From Physiology To Clinical Implications: Acid-Base, Electrolyte and Fluid Alterations: ReviewDokument8 SeitenPotassium: From Physiology To Clinical Implications: Acid-Base, Electrolyte and Fluid Alterations: Reviewkarlakalua77Noch keine Bewertungen
- Faculty Version With Model Answers: Body FluidsDokument10 SeitenFaculty Version With Model Answers: Body FluidsSoji AdimulaNoch keine Bewertungen
- An Introduction To Gas Chromatography-Mass Spectrometry and The Inherited Organic AcidemiasDokument12 SeitenAn Introduction To Gas Chromatography-Mass Spectrometry and The Inherited Organic AcidemiasFefe SaiedNoch keine Bewertungen
- Baker 1954Dokument35 SeitenBaker 1954Fatima AhmedNoch keine Bewertungen
- STPM Chapter 3 ExercisesDokument8 SeitenSTPM Chapter 3 ExercisesVashniie TabithaNoch keine Bewertungen
- ABG Made Easy FinalDokument144 SeitenABG Made Easy FinalsurasuarezlopezNoch keine Bewertungen
- STD - XII Subject: Chemistry:) 1l!9.hyck9JIDokument6 SeitenSTD - XII Subject: Chemistry:) 1l!9.hyck9JISaktheeswaran VNoch keine Bewertungen
- Boerhaavia Diffusa Hepatoprotective PDFDokument9 SeitenBoerhaavia Diffusa Hepatoprotective PDFShivani KalokheNoch keine Bewertungen
- GFR RBF Tubular in Infancy 1948Dokument9 SeitenGFR RBF Tubular in Infancy 1948Nia Prajnya SyailendraNoch keine Bewertungen
- 1721BEEE30083 EE701-N IPS 14 Sept 2020Dokument3 Seiten1721BEEE30083 EE701-N IPS 14 Sept 2020Ludwig BakedtheovenNoch keine Bewertungen
- Diseases: Intercalated Cells: More Than PH RegulationDokument22 SeitenDiseases: Intercalated Cells: More Than PH RegulationIvanNoch keine Bewertungen
- Counter Current Gas-Liquid Vertical Flow Model For Flow Pattern and Pressure Drop, Taitel and BarneaDokument11 SeitenCounter Current Gas-Liquid Vertical Flow Model For Flow Pattern and Pressure Drop, Taitel and Barneapablo raffinNoch keine Bewertungen
- Zinc CortisolDokument7 SeitenZinc CortisoldocumentosdescribdNoch keine Bewertungen
- Fisio 1Dokument11 SeitenFisio 1anaNoch keine Bewertungen
- Bab I PendahuluanDokument43 SeitenBab I Pendahuluanpaklay2Noch keine Bewertungen
- Glycosuric DiuresisDokument12 SeitenGlycosuric DiuresisNida NazimNoch keine Bewertungen
- Arab Board 2017 Internal MedicineDokument37 SeitenArab Board 2017 Internal MedicineAFAQ MAMMEDZADENoch keine Bewertungen
- Plasma CalciumDokument4 SeitenPlasma CalciumÁureo BarbosaNoch keine Bewertungen
- Control Gasçflowrate OrificeDokument11 SeitenControl Gasçflowrate Orificepma61Noch keine Bewertungen
- Mathematics Lab Manual (Activity 1)Dokument5 SeitenMathematics Lab Manual (Activity 1)Heeral SinghNoch keine Bewertungen
- The Preparation and Function of The Hypertensin-Converting EnzymeDokument5 SeitenThe Preparation and Function of The Hypertensin-Converting EnzymeDana MNoch keine Bewertungen
- 09 - Chapter 2Dokument18 Seiten09 - Chapter 2bijesh babuNoch keine Bewertungen
- Brief Communication: Oxyradicals and Multivitamin TabletsDokument2 SeitenBrief Communication: Oxyradicals and Multivitamin TabletsfsdfsNoch keine Bewertungen
- 1 Johor QDokument22 Seiten1 Johor QLewin HongNoch keine Bewertungen
- 1977 - The Mellanby Effect On Moderate and Heavy DrinkerDokument6 Seiten1977 - The Mellanby Effect On Moderate and Heavy DrinkerIsnia FitrasantiNoch keine Bewertungen
- 2012 Dec P1 (Arab Board)Dokument26 Seiten2012 Dec P1 (Arab Board)Mustafa Ismael NayyefNoch keine Bewertungen
- Studies On The Swimming Musculature of The Rainbow Trout. II. Muscle Metabolism During Severe HypoxiaDokument9 SeitenStudies On The Swimming Musculature of The Rainbow Trout. II. Muscle Metabolism During Severe HypoxiaLopez IvanNoch keine Bewertungen
- Tutoral 3 Solution PDFDokument11 SeitenTutoral 3 Solution PDFBenny ChristianNoch keine Bewertungen
- Chemical Reaction Fundimantal For Chemical Engineering - Part339Dokument1 SeiteChemical Reaction Fundimantal For Chemical Engineering - Part339tysir sarhanNoch keine Bewertungen
- Revenue CodeDokument110 SeitenRevenue CodemargemallowsNoch keine Bewertungen
- J. Biol. Chem.-1943-Stark-319-25Dokument8 SeitenJ. Biol. Chem.-1943-Stark-319-25gibrambo5770Noch keine Bewertungen
- Hemoglobin ConcentrationDokument19 SeitenHemoglobin ConcentrationDominique ReyesNoch keine Bewertungen
- Construir - FermentadorDokument6 SeitenConstruir - FermentadorROBERTO JHALVER VEGA PAULINONoch keine Bewertungen
- Well Test Previous Year QuestionDokument13 SeitenWell Test Previous Year QuestionDiptoNoch keine Bewertungen
- P Silo Cyn ExtractionDokument4 SeitenP Silo Cyn ExtractionSmokeNoch keine Bewertungen
- Adobe Scan 26 Apr 2023Dokument1 SeiteAdobe Scan 26 Apr 2023Sahil ShindeNoch keine Bewertungen
- R. Dawson and Keiko Tokuyama: Department Chemistry, Columbia University, York, N.YDokument11 SeitenR. Dawson and Keiko Tokuyama: Department Chemistry, Columbia University, York, N.YVinaFitrianiPratiwiNoch keine Bewertungen
- Biotech Bioengineering - March 1976 - Verhoff - Mass and Energy Balance Analysis of Metabolic Pathways Applied To CitricDokument8 SeitenBiotech Bioengineering - March 1976 - Verhoff - Mass and Energy Balance Analysis of Metabolic Pathways Applied To CitricTamirat kebedeNoch keine Bewertungen
- RLP: Il (T: VQ:: Phieij Chinh Ro-Le TramDokument7 SeitenRLP: Il (T: VQ:: Phieij Chinh Ro-Le TramTHANHDIEPNoch keine Bewertungen
- 1ST Prof Physio PaperDokument2 Seiten1ST Prof Physio Papermadeha goharNoch keine Bewertungen
- Chap 12Dokument17 SeitenChap 12Tai DonovanNoch keine Bewertungen
- Animal Anatomy and Physiology: Figure Q.18Dokument25 SeitenAnimal Anatomy and Physiology: Figure Q.18hager atefNoch keine Bewertungen
- 10.1016@0009 89816890264 7Dokument5 Seiten10.1016@0009 89816890264 7Puku KunNoch keine Bewertungen
- Speed Surgery SsDokument20 SeitenSpeed Surgery SsPavan KumarNoch keine Bewertungen
- Votage: B) Defime Cutreut, Voltage and Eergy-3MDokument12 SeitenVotage: B) Defime Cutreut, Voltage and Eergy-3MPaybackNoch keine Bewertungen
- Ormood Aits ,: Tory Failure Reaso, As For Which May Be The Primary ReasonDokument6 SeitenOrmood Aits ,: Tory Failure Reaso, As For Which May Be The Primary ReasonTiaRa JuraidNoch keine Bewertungen
- Acute Pancreatitis Due To Pancreatic Hydatid Cyst: A Case Report and Review of The Literature Amin MakniDokument18 SeitenAcute Pancreatitis Due To Pancreatic Hydatid Cyst: A Case Report and Review of The Literature Amin MaknifentyertantiNoch keine Bewertungen
- The Living WorldDokument10 SeitenThe Living WorldShankar MurmuNoch keine Bewertungen
- Diaz GJ, Perilla NS and Rojas YDokument6 SeitenDiaz GJ, Perilla NS and Rojas YKelin GallardoNoch keine Bewertungen
- The Effect of Systox On Ionic Fluxes in Excised Barley RootsDokument10 SeitenThe Effect of Systox On Ionic Fluxes in Excised Barley RootsSh1vaNoch keine Bewertungen
- Effect of Saliva On Sperm Motility and Activity: Togas Tulandi, M.D. Leo Plouffe, JR., M.D.T Robert Mcinnes, M.DDokument3 SeitenEffect of Saliva On Sperm Motility and Activity: Togas Tulandi, M.D. Leo Plouffe, JR., M.D.T Robert Mcinnes, M.DWesley Faruk ANoch keine Bewertungen
- Fluid TherapiDokument158 SeitenFluid TherapiAsmalina AzizanNoch keine Bewertungen
- The SGOT/SGPT Ratio - An Indicator of Alcoholic Liver DiseaseDokument4 SeitenThe SGOT/SGPT Ratio - An Indicator of Alcoholic Liver DiseaseSahara MaindokaNoch keine Bewertungen
- Orthopaedic X RaysDokument4 SeitenOrthopaedic X Raysdarsh bhattiNoch keine Bewertungen
- The Structure of The ChloritesDokument7 SeitenThe Structure of The ChloritesKaram JaradatNoch keine Bewertungen
- Asphyxia Case StudyDokument8 SeitenAsphyxia Case StudySanny Ramos100% (2)
- Altered ThermoregulationDokument29 SeitenAltered Thermoregulationsreekala100% (1)
- Pulse DiagnoseDokument427 SeitenPulse DiagnoseSnezana MihajlovicNoch keine Bewertungen
- Ocurest Plus2018Dokument4 SeitenOcurest Plus2018rotastrainNoch keine Bewertungen
- PharmacologyDokument236 SeitenPharmacologynbde2100% (5)
- 1regional BlockDokument150 Seiten1regional BlockLilija SöderholmNoch keine Bewertungen
- Fluid Therapy and The Microcirculation in Health and Critical IllnessDokument11 SeitenFluid Therapy and The Microcirculation in Health and Critical IllnessAdote DrmNoch keine Bewertungen
- Adrenergic DrugsDokument2 SeitenAdrenergic DrugsSunshine_Bacla_4275Noch keine Bewertungen
- BIO271 - Practice MCQDokument6 SeitenBIO271 - Practice MCQEric LuongNoch keine Bewertungen
- 2014 Article 390 PDFDokument8 Seiten2014 Article 390 PDFGlucose DRglucoseNoch keine Bewertungen
- Makalah Bahasa Inggris Stik AvicennaDokument16 SeitenMakalah Bahasa Inggris Stik AvicennahemraninNoch keine Bewertungen
- @anesthesia - Books 2015 MCQs For Handbook of Local Anesthesia PDFDokument205 Seiten@anesthesia - Books 2015 MCQs For Handbook of Local Anesthesia PDFVivek Sinha100% (2)
- Stone Therapy Manual Guild1Dokument46 SeitenStone Therapy Manual Guild1Envie AestheticsNoch keine Bewertungen
- Cardiovascular Drift and Cerebral and Muscle Tissue Oxygenation During Prolonged Cycling at Different Pedalling CadencesDokument12 SeitenCardiovascular Drift and Cerebral and Muscle Tissue Oxygenation During Prolonged Cycling at Different Pedalling CadencesBob UeckerleleNoch keine Bewertungen
- Understanding HemodynamicsDokument21 SeitenUnderstanding HemodynamicsGiovanni Mictil86% (7)
- Pathophysiology of Hydrops FetalisDokument10 SeitenPathophysiology of Hydrops FetalisdrsunilpawarNoch keine Bewertungen
- A&P Cardiovascular System PowerPoint (Nursing)Dokument34 SeitenA&P Cardiovascular System PowerPoint (Nursing)Linsey Bowen67% (6)
- 7.effects of Sympathetic and Parasympathetic Stimulation On SpecificDokument16 Seiten7.effects of Sympathetic and Parasympathetic Stimulation On SpecificSanthoshi Sadhanaa SankarNoch keine Bewertungen
- Pediatric Endocrinology A Practical Clinical Guide, 2E 2013 (PDF) (DR - Carson) VRGDokument263 SeitenPediatric Endocrinology A Practical Clinical Guide, 2E 2013 (PDF) (DR - Carson) VRGIonela RobertaNoch keine Bewertungen
- Ciclo Nasal RespiratorioDokument7 SeitenCiclo Nasal RespiratorioManuel RaGalNoch keine Bewertungen
- Local Anesthesia Part-1Dokument33 SeitenLocal Anesthesia Part-1Abdul RazakNoch keine Bewertungen
- Ice TherapyDokument5 SeitenIce TherapyDiane CastillonNoch keine Bewertungen
- NCP For HypertensionDokument6 SeitenNCP For HypertensionJaic Ealston D. Tampus0% (2)
- J Survophthal 2018 10 005Dokument28 SeitenJ Survophthal 2018 10 005Luis LondoñoNoch keine Bewertungen
- Drugs Acting On Cardiovascular SystemDokument18 SeitenDrugs Acting On Cardiovascular SystemIbrahem AlNoch keine Bewertungen
- ADRENERGIC RECEPTORS-Leraning Objectives & NotesDokument10 SeitenADRENERGIC RECEPTORS-Leraning Objectives & NotesManikanta GupthaNoch keine Bewertungen
- WawaDokument199 SeitenWawacolteapaulNoch keine Bewertungen
- Hypertension PDFDokument50 SeitenHypertension PDFMochamad Ali Rosadi100% (1)
- Effects of HypoxiaDokument6 SeitenEffects of HypoxiaMaria TelloNoch keine Bewertungen
- Arterial Blood PressureDokument46 SeitenArterial Blood PressureRupasi KathiravanNoch keine Bewertungen
- Power of 10: The Once-A-Week Slow Motion Fitness RevolutionVon EverandPower of 10: The Once-A-Week Slow Motion Fitness RevolutionBewertung: 3.5 von 5 Sternen3.5/5 (11)
- Peak: The New Science of Athletic Performance That is Revolutionizing SportsVon EverandPeak: The New Science of Athletic Performance That is Revolutionizing SportsBewertung: 5 von 5 Sternen5/5 (97)
- Chair Yoga: Sit, Stretch, and Strengthen Your Way to a Happier, Healthier YouVon EverandChair Yoga: Sit, Stretch, and Strengthen Your Way to a Happier, Healthier YouBewertung: 3.5 von 5 Sternen3.5/5 (5)
- Boundless: Upgrade Your Brain, Optimize Your Body & Defy AgingVon EverandBoundless: Upgrade Your Brain, Optimize Your Body & Defy AgingBewertung: 4.5 von 5 Sternen4.5/5 (67)
- Strong Is the New Beautiful: Embrace Your Natural Beauty, Eat Clean, and Harness Your PowerVon EverandStrong Is the New Beautiful: Embrace Your Natural Beauty, Eat Clean, and Harness Your PowerBewertung: 4 von 5 Sternen4/5 (5)
- Relentless: From Good to Great to UnstoppableVon EverandRelentless: From Good to Great to UnstoppableBewertung: 5 von 5 Sternen5/5 (787)
- Aging Backwards: Reverse the Aging Process and Look 10 Years Younger in 30 Minutes a DayVon EverandAging Backwards: Reverse the Aging Process and Look 10 Years Younger in 30 Minutes a DayNoch keine Bewertungen
- The Total Kettlebell Workout: Trade Secrets of a Personal TrainerVon EverandThe Total Kettlebell Workout: Trade Secrets of a Personal TrainerBewertung: 5 von 5 Sternen5/5 (1)
- The Yogi Code: Seven Universal Laws of Infinite SuccessVon EverandThe Yogi Code: Seven Universal Laws of Infinite SuccessBewertung: 4.5 von 5 Sternen4.5/5 (104)
- Pranayama: The Yoga Science of BreathingVon EverandPranayama: The Yoga Science of BreathingBewertung: 4.5 von 5 Sternen4.5/5 (8)
- Muscle for Life: Get Lean, Strong, and Healthy at Any Age!Von EverandMuscle for Life: Get Lean, Strong, and Healthy at Any Age!Bewertung: 4.5 von 5 Sternen4.5/5 (22)
- Strength Training Over 40: The Only Weight Training Workout Book You Will Need to Maintain or Build Your Strength, Muscle Mass, Energy, Overall Fitness and Stay Healthy Without Living in the GymVon EverandStrength Training Over 40: The Only Weight Training Workout Book You Will Need to Maintain or Build Your Strength, Muscle Mass, Energy, Overall Fitness and Stay Healthy Without Living in the GymBewertung: 4 von 5 Sternen4/5 (6)
- Functional Training and Beyond: Building the Ultimate Superfunctional Body and MindVon EverandFunctional Training and Beyond: Building the Ultimate Superfunctional Body and MindBewertung: 4.5 von 5 Sternen4.5/5 (1)
- The Power of Fastercise: Using the New Science of Signaling Exercise to Get Surprisingly Fit in Just a Few Minutes a DayVon EverandThe Power of Fastercise: Using the New Science of Signaling Exercise to Get Surprisingly Fit in Just a Few Minutes a DayBewertung: 4 von 5 Sternen4/5 (4)
- Mind Your Body: 4 Weeks to a Leaner, Healthier LifeVon EverandMind Your Body: 4 Weeks to a Leaner, Healthier LifeBewertung: 4.5 von 5 Sternen4.5/5 (5)
- The Yoga Sutras of Patanjali: The Final Guide for the Study and Practice of Patanjali's Yoga SutrasVon EverandThe Yoga Sutras of Patanjali: The Final Guide for the Study and Practice of Patanjali's Yoga SutrasNoch keine Bewertungen
- Easy Strength: How to Get a Lot Stronger Than Your Competition-And Dominate in Your SportVon EverandEasy Strength: How to Get a Lot Stronger Than Your Competition-And Dominate in Your SportBewertung: 4.5 von 5 Sternen4.5/5 (17)
- 80/20 Running: Run Stronger and Race Faster by Training SlowerVon Everand80/20 Running: Run Stronger and Race Faster by Training SlowerBewertung: 4.5 von 5 Sternen4.5/5 (97)
- Yamas & Niyamas: Exploring Yoga's Ethical PracticeVon EverandYamas & Niyamas: Exploring Yoga's Ethical PracticeBewertung: 4.5 von 5 Sternen4.5/5 (111)
- Endure: Mind, Body, and the Curiously Elastic Limits of Human PerformanceVon EverandEndure: Mind, Body, and the Curiously Elastic Limits of Human PerformanceBewertung: 4.5 von 5 Sternen4.5/5 (237)
- Calisthenics: Guide for Bodyweight Exercise, Build your Dream Body in 30 MinutesVon EverandCalisthenics: Guide for Bodyweight Exercise, Build your Dream Body in 30 MinutesBewertung: 3 von 5 Sternen3/5 (5)
- Not a Diet Book: Take Control. Gain Confidence. Change Your Life.Von EverandNot a Diet Book: Take Control. Gain Confidence. Change Your Life.Bewertung: 4.5 von 5 Sternen4.5/5 (124)
- Move Your DNA 2nd ed: Restore Your Health Through Natural MovementVon EverandMove Your DNA 2nd ed: Restore Your Health Through Natural MovementBewertung: 4 von 5 Sternen4/5 (8)
- Enter The Kettlebell!: Strength Secret of the Soviet SupermenVon EverandEnter The Kettlebell!: Strength Secret of the Soviet SupermenBewertung: 4 von 5 Sternen4/5 (29)
- Roxane Gay & Everand Originals: Built for This: The Quiet Strength of PowerliftingVon EverandRoxane Gay & Everand Originals: Built for This: The Quiet Strength of PowerliftingBewertung: 4.5 von 5 Sternen4.5/5 (133)
- 8 Weeks to 30 Consecutive Pull-Ups: Build Your Upper Body Working Your Upper Back, Shoulders, and Biceps | at Home Workouts | No Gym Required |Von Everand8 Weeks to 30 Consecutive Pull-Ups: Build Your Upper Body Working Your Upper Back, Shoulders, and Biceps | at Home Workouts | No Gym Required |Noch keine Bewertungen