Das könnte Ihnen auch gefallen
- Prontuario NR 20Dokument12 SeitenProntuario NR 20Thiago Jacomo50% (2)
- Aula 2 Validação de ProcessosDokument60 SeitenAula 2 Validação de Processosgabriele100% (1)
- Aula HVAC - 15 - 03 - 2022 - RacineDokument75 SeitenAula HVAC - 15 - 03 - 2022 - RacineQualidade Mag IndustriaNoch keine Bewertungen
- Manual Saneantes PDFDokument165 SeitenManual Saneantes PDFNorma FerreiraNoch keine Bewertungen
- Abnt NBR 15450Dokument19 SeitenAbnt NBR 15450Jorge CiprianoNoch keine Bewertungen
- Validação de Sistemas Computadorizados - 15 e 16-12-2020 - PBDokument232 SeitenValidação de Sistemas Computadorizados - 15 e 16-12-2020 - PBFrank MiguelNoch keine Bewertungen
- Abnt NBR 16035-1Dokument25 SeitenAbnt NBR 16035-1Marcosbss100% (3)
- Aula 14 - Validação de ProcessosDokument138 SeitenAula 14 - Validação de ProcessosSuzana BenderNoch keine Bewertungen
- Procedimento para Controle de Dispositivos de Medição e MonitoramentoDokument22 SeitenProcedimento para Controle de Dispositivos de Medição e MonitoramentoTiago A. Silva0% (1)
- Qualificação X CalibraçãoDokument62 SeitenQualificação X CalibraçãobarneyshowNoch keine Bewertungen
- PQ 10 Monitorização Medição ProdutoDokument2 SeitenPQ 10 Monitorização Medição ProdutoFabio Dantas100% (1)
- Ordem de ServiçoDokument2 SeitenOrdem de ServiçoNestor W Neto83% (6)
- Relatorio de Torqueamento de Parafusos Rev 00Dokument1 SeiteRelatorio de Torqueamento de Parafusos Rev 00Abraham Silva100% (2)
- Validação de Equipamentos em CME - Luciano ManoelDokument54 SeitenValidação de Equipamentos em CME - Luciano ManoelRafael Dias100% (1)
- Ergonomia Aula Pratica PDFDokument22 SeitenErgonomia Aula Pratica PDFdiegoNoch keine Bewertungen
- Abnt - NBR 12966 - Avaliacao Tecnica de Fornecedores PDFDokument6 SeitenAbnt - NBR 12966 - Avaliacao Tecnica de Fornecedores PDFDeco DluxeNoch keine Bewertungen
- NIT Dicla 12 - 20 PDFDokument75 SeitenNIT Dicla 12 - 20 PDFGabriel Meireles100% (1)
- Arte Eletronica de Potencia PDFDokument130 SeitenArte Eletronica de Potencia PDFThiago NunesNoch keine Bewertungen
- Apresentação Lapidação 01Dokument60 SeitenApresentação Lapidação 01Solon Gomes Amaral100% (1)
- 07 - Planos de Controle e Monitoramento AmbientalDokument3 Seiten07 - Planos de Controle e Monitoramento AmbientalWellik SouzaNoch keine Bewertungen
- Gestão de EquipamentosDokument70 SeitenGestão de EquipamentosJizreel Cristo100% (1)
- Guias de QualidadeDokument61 SeitenGuias de Qualidadepessoathiago100% (1)
- Comparação Da RDC 210 X 17Dokument40 SeitenComparação Da RDC 210 X 17Jonas Rafael Pereira MartinsNoch keine Bewertungen
- An06 - Requisitos Gerais Inspeção FabricaçãoDokument10 SeitenAn06 - Requisitos Gerais Inspeção FabricaçãoGLEDSONNoch keine Bewertungen
- Aula 4 - Qualificação e Validação Na Indústria de Medicamentos V2Dokument28 SeitenAula 4 - Qualificação e Validação Na Indústria de Medicamentos V2Larissa DuarteNoch keine Bewertungen
- IN Nº138 - Boas Práticas de Fabricação Complementares Às Atividades de Qualificação e ValidaçãoDokument24 SeitenIN Nº138 - Boas Práticas de Fabricação Complementares Às Atividades de Qualificação e ValidaçãoBrunna Schuch VilanovaNoch keine Bewertungen
- GP - PQ - AracruzDokument4 SeitenGP - PQ - AracruzLisianeNoch keine Bewertungen
- CURSO - RDC 16 Apresentação - PPT - REV Jan 2021Dokument200 SeitenCURSO - RDC 16 Apresentação - PPT - REV Jan 2021Mendes Consultorias100% (3)
- Workshop RDC 48 Marcia Palomares Fasb GARANTIA DA QUALIDADEDokument11 SeitenWorkshop RDC 48 Marcia Palomares Fasb GARANTIA DA QUALIDADErobinquimicaNoch keine Bewertungen
- MinutaDokument7 SeitenMinutaSilva ErreniteNoch keine Bewertungen
- 04-18-Documentação Técnica e Registros Da Qualidade - 6pgsDokument6 Seiten04-18-Documentação Técnica e Registros Da Qualidade - 6pgspablo da silva oliveiraNoch keine Bewertungen
- ABC Da InspeçãoDokument24 SeitenABC Da InspeçãoIgor SampaioNoch keine Bewertungen
- Curso Intermediário Monsanto - Nr20 Parte 3Dokument58 SeitenCurso Intermediário Monsanto - Nr20 Parte 3Adriano Loureiro De SouzaNoch keine Bewertungen
- PGCP-118 A - Procedimento - Geral - para - Certificacao - de - ProdutosDokument43 SeitenPGCP-118 A - Procedimento - Geral - para - Certificacao - de - ProdutosDaniel Jorge Da SilvaNoch keine Bewertungen
- E Book AnatelDokument25 SeitenE Book AnatelMaria Cecilia Basilio RibeiroNoch keine Bewertungen
- RDC #658Dokument17 SeitenRDC #658brunaemilialimaNoch keine Bewertungen
- Diário Oficial Da União: Instrução Normativa in #138, de 30 de Março de 2022Dokument14 SeitenDiário Oficial Da União: Instrução Normativa in #138, de 30 de Março de 2022Robson CarneiroNoch keine Bewertungen
- Quais São As Boas Práticas de FabricaçãoDokument2 SeitenQuais São As Boas Práticas de FabricaçãolidyaniNoch keine Bewertungen
- In 134-2022Dokument9 SeitenIn 134-2022Paulo Alberto Gomes Silva HartmannNoch keine Bewertungen
- Regimento Geral Do SIAC e AnexosDokument106 SeitenRegimento Geral Do SIAC e AnexosZara CorretoraNoch keine Bewertungen
- Diário Oficial Da União: Instrução Normativa in #134, de 30 de Março de 2022Dokument6 SeitenDiário Oficial Da União: Instrução Normativa in #134, de 30 de Março de 2022Mauro SilvaNoch keine Bewertungen
- RGI - Regulamento Geral de InspeçãoDokument14 SeitenRGI - Regulamento Geral de InspeçãoFabricio RabeloNoch keine Bewertungen
- Anexo I - Guia de Auditoria - Fornecedor - Rev4-17609824Dokument7 SeitenAnexo I - Guia de Auditoria - Fornecedor - Rev4-17609824Eric DuarteNoch keine Bewertungen
- 9 P-03 Parte I - Odilon HortaDokument65 Seiten9 P-03 Parte I - Odilon HortaLincoln TCNoch keine Bewertungen
- FT 16 e 17 - Sistema APPCC - Análise de Perigos e Pontos Críticos de Controle - GQ 13056-1Dokument5 SeitenFT 16 e 17 - Sistema APPCC - Análise de Perigos e Pontos Críticos de Controle - GQ 13056-1janio araujo mouraNoch keine Bewertungen
- NTC-003-06 - Proced - Diligência, Amostra e Inspeção de ProdutoDokument12 SeitenNTC-003-06 - Proced - Diligência, Amostra e Inspeção de ProdutoLeandro SilvaNoch keine Bewertungen
- 118-2015 - RGCPDokument46 Seiten118-2015 - RGCPNelson CoelhoNoch keine Bewertungen
- In 138 2022Dokument22 SeitenIn 138 2022Maria Júlia Lovo de FreitasNoch keine Bewertungen
- Apresentação Metrologia Parte 4Dokument14 SeitenApresentação Metrologia Parte 4Jean Sarmento FerrazNoch keine Bewertungen
- Abnt NBR Iso Iec 17025 2005 PDFDokument33 SeitenAbnt NBR Iso Iec 17025 2005 PDFJonas AriclenesNoch keine Bewertungen
- Inmetro Portaria 457 de 2010 - RGCPDokument29 SeitenInmetro Portaria 457 de 2010 - RGCPBruno BritoNoch keine Bewertungen
- Cartilha GMP A4Dokument2 SeitenCartilha GMP A4Andrea Carignani BensadonNoch keine Bewertungen
- Avaliação Da Conformidade - Requisitos para Organismos de Certificação de Produtos, Processos e ServiçosDokument41 SeitenAvaliação Da Conformidade - Requisitos para Organismos de Certificação de Produtos, Processos e ServiçosNeemias AlmeidaNoch keine Bewertungen
- Instrução Normativa - in #47, de 21 de Agosto de 2019 - Qualificação e ValidaçãoDokument14 SeitenInstrução Normativa - in #47, de 21 de Agosto de 2019 - Qualificação e ValidaçãoAlan CarvalhoNoch keine Bewertungen
- INSTRUÇÃO NORMATIVA IN #134, DE 30 DE Março DE 2022 - INSTRUÇÃO NORMATIVA IN #134, DE 30 DE Março DE 2022 - DOU - Imprensa NacionalDokument6 SeitenINSTRUÇÃO NORMATIVA IN #134, DE 30 DE Março DE 2022 - INSTRUÇÃO NORMATIVA IN #134, DE 30 DE Março DE 2022 - DOU - Imprensa NacionalLucas SilvaNoch keine Bewertungen
- RDC 11 2012Dokument12 SeitenRDC 11 2012spirotessNoch keine Bewertungen
- INSTRUÇÃO NORMATIVA - IN #43, DE 21 DE AGOSTO DE 2019 - Instrucao Sobre Topicos de ValidaçãoDokument6 SeitenINSTRUÇÃO NORMATIVA - IN #43, DE 21 DE AGOSTO DE 2019 - Instrucao Sobre Topicos de ValidaçãoluizpirezNoch keine Bewertungen
- NIT Dicor 1 - 21Dokument14 SeitenNIT Dicor 1 - 21Samantha MurilloNoch keine Bewertungen
- RDC 17 ResumoDokument10 SeitenRDC 17 ResumoLucas Souza Dos Santos100% (1)
- NA-012 Rev. 04 (Jun-2021) Qualif. e Certif. de Pessoas em Insp. de FabricaçãoDokument45 SeitenNA-012 Rev. 04 (Jun-2021) Qualif. e Certif. de Pessoas em Insp. de FabricaçãodionepsouzaNoch keine Bewertungen
- 12 - Colettania IF-N1-CTDokument58 Seiten12 - Colettania IF-N1-CTAntonio CarlosNoch keine Bewertungen
- Regimento QualificacaoDokument99 SeitenRegimento QualificacaoDiego AguiarNoch keine Bewertungen
- Nit Dicor 65 - 01Dokument17 SeitenNit Dicor 65 - 01Renan PiaiNoch keine Bewertungen
- Portaria 327 BPFC - Produtos SanitizantesDokument15 SeitenPortaria 327 BPFC - Produtos SanitizantesRaison BusinaroNoch keine Bewertungen
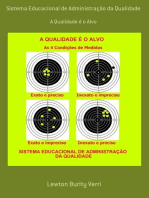
- Sistema Educacional De Administração Da QualidadeVon EverandSistema Educacional De Administração Da QualidadeNoch keine Bewertungen
- CP #64 GgtesDokument15 SeitenCP #64 GgtesRafael DiasNoch keine Bewertungen
- Farmácias de ManipulaçãoDokument15 SeitenFarmácias de ManipulaçãoRafael DiasNoch keine Bewertungen
- Manual Ar Condicionado AAAAAA PDFDokument24 SeitenManual Ar Condicionado AAAAAA PDFRafael DiasNoch keine Bewertungen
- Revista PortfolioDokument44 SeitenRevista PortfolioRafael Dias100% (1)
- Análise Financeira Geral 2019 - Compressed 1Dokument48 SeitenAnálise Financeira Geral 2019 - Compressed 1Rafael DiasNoch keine Bewertungen
- Art e Cat - ConfeaDokument7 SeitenArt e Cat - ConfeaRafael DiasNoch keine Bewertungen
- Abnt-nbr-16035-1-Caldeiras e Vasos de Pressão - Requisitos Mínimos para A ConstruçãoDokument2 SeitenAbnt-nbr-16035-1-Caldeiras e Vasos de Pressão - Requisitos Mínimos para A ConstruçãoRafael DiasNoch keine Bewertungen
- RDC 17-2010 BPFDokument109 SeitenRDC 17-2010 BPFCarla PetersNoch keine Bewertungen
- Resolução Conjunta 9 447 de 15 12 15 Pesquisa de PreçosDokument6 SeitenResolução Conjunta 9 447 de 15 12 15 Pesquisa de PreçosRafael DiasNoch keine Bewertungen
- Apostila Msa PortuguêsDokument28 SeitenApostila Msa PortuguêsmsbarretosNoch keine Bewertungen
- RDC17 - 2010c I Legislação Indústria FarmacêuticaDokument32 SeitenRDC17 - 2010c I Legislação Indústria FarmacêuticaVivian KusanoNoch keine Bewertungen
- Registro Pessoa Juridica - ConfeaDokument3 SeitenRegistro Pessoa Juridica - ConfeaRafael DiasNoch keine Bewertungen
- Conhecendo O Processo Produtivo de Sólidos, Líquidos E Granulação Na Indústria FarmacêuticaDokument31 SeitenConhecendo O Processo Produtivo de Sólidos, Líquidos E Granulação Na Indústria FarmacêuticaRafael DiasNoch keine Bewertungen
- EmpréstimoDokument4 SeitenEmpréstimoRafael DiasNoch keine Bewertungen
- Apostila Acionamentos Eletricos 2008 NeemiasDokument56 SeitenApostila Acionamentos Eletricos 2008 Neemiasrodrigo_0909Noch keine Bewertungen
- Bomba de Teste para Produzir Pressão de Ensaio - Bomba de Teste para o Kit de Baixa de PressãoDokument1 SeiteBomba de Teste para Produzir Pressão de Ensaio - Bomba de Teste para o Kit de Baixa de PressãoRafael DiasNoch keine Bewertungen
- Artigo Produção de ComprimidosDokument9 SeitenArtigo Produção de ComprimidosDiego GifoniNoch keine Bewertungen
- 01 Msa ApresentacaoDokument66 Seiten01 Msa ApresentacaoAlecir SilvaNoch keine Bewertungen
- Graficos de Controle No ExcelDokument16 SeitenGraficos de Controle No ExcelRafael DiasNoch keine Bewertungen
- Produção MedDokument37 SeitenProdução MedRafael DiasNoch keine Bewertungen
- Nota Técnica Anemometro FinalDokument1 SeiteNota Técnica Anemometro FinalRafael DiasNoch keine Bewertungen
- DOQ Cgcre 36 - 00Dokument7 SeitenDOQ Cgcre 36 - 00Isabel LimaNoch keine Bewertungen
- Semana 3Dokument33 SeitenSemana 3Eliane CristinaNoch keine Bewertungen
- Cartilha RobertoGarciaDokument26 SeitenCartilha RobertoGarciarobert santosNoch keine Bewertungen
- Manual Técnico Incubadora SCTI Line 4 V3Dokument48 SeitenManual Técnico Incubadora SCTI Line 4 V3Clodoaldo BorgesNoch keine Bewertungen
- CE5000 ManualDokument122 SeitenCE5000 ManualEmerson SilvaNoch keine Bewertungen
- TCC-versãofinal2 1Dokument52 SeitenTCC-versãofinal2 1Alex LopesNoch keine Bewertungen
- CST Csosn PDFDokument1 SeiteCST Csosn PDFLuciano Dos SantosNoch keine Bewertungen
- Assinando Digitalmente Um XML Usando C# - Profissionais TI - Pra Quem Respira InformaçãoDokument12 SeitenAssinando Digitalmente Um XML Usando C# - Profissionais TI - Pra Quem Respira InformaçãoIvanilson ContabilidadeNoch keine Bewertungen
- Es - DT.PDN.02.01.006 - Construção e ManutençãoDokument14 SeitenEs - DT.PDN.02.01.006 - Construção e ManutençãoGabriel WelsingNoch keine Bewertungen
- ACO Documentos de InspecaoDokument2 SeitenACO Documentos de Inspecaogf__8272Noch keine Bewertungen
- APO2023 - ED9 - Cap18 e 20 - ResolvidoDokument7 SeitenAPO2023 - ED9 - Cap18 e 20 - ResolvidoCesar SampaioNoch keine Bewertungen
- Barão de Mauá II - Regulamento Interno 040723Dokument24 SeitenBarão de Mauá II - Regulamento Interno 040723Adalberto LucioNoch keine Bewertungen
- Técnicas Construtivas Modernas Na Construção CivilDokument9 SeitenTécnicas Construtivas Modernas Na Construção CivilwhallefNoch keine Bewertungen
- Controle de Vibrações Geradas Por Desmonte de Rocha Com Explosivos. Estudo de Caso - Calcário Cruzeiro, Limeira (SP)Dokument12 SeitenControle de Vibrações Geradas Por Desmonte de Rocha Com Explosivos. Estudo de Caso - Calcário Cruzeiro, Limeira (SP)daniel_tavares_9Noch keine Bewertungen
- IsoDokument49 SeitenIsoAndressaNunesNoch keine Bewertungen
- Divulgação de Resultados: Prévia Operacional 1T22Dokument27 SeitenDivulgação de Resultados: Prévia Operacional 1T22andre.torresNoch keine Bewertungen
- Operação e Manutenção de Sistemas de Abastecimento de ÁguaDokument4 SeitenOperação e Manutenção de Sistemas de Abastecimento de ÁguaRaphael Fagundes LopesNoch keine Bewertungen
- Mala DiretaDokument2 SeitenMala DiretaArlete LetyNoch keine Bewertungen
- Relatório Final - Projeto Fapesp - Uso de Piezoelétricos para Flow Control - Com Capa Sem AssinarDokument34 SeitenRelatório Final - Projeto Fapesp - Uso de Piezoelétricos para Flow Control - Com Capa Sem AssinarDanilo AlarconNoch keine Bewertungen
- NBRNM Iso3842Dokument15 SeitenNBRNM Iso3842JUNIORNoch keine Bewertungen
- PES - Esquadrias - Batente e PortaDokument6 SeitenPES - Esquadrias - Batente e PortaAlexandre CostaNoch keine Bewertungen
- Resumo Nao Tecnico - 29022016Dokument19 SeitenResumo Nao Tecnico - 29022016Luís CastroNoch keine Bewertungen
- Catalogo JeeneDokument32 SeitenCatalogo JeeneUser AdmNoch keine Bewertungen
- LE503 - Tecnologia Mecânica - Introdução PDFDokument23 SeitenLE503 - Tecnologia Mecânica - Introdução PDFRodrigo ContieriNoch keine Bewertungen
- Associação de ResistoresDokument13 SeitenAssociação de ResistoresLeonardo MardeganNoch keine Bewertungen