Das könnte Ihnen auch gefallen
- Cleanroom Technology: Fundamentals of Design, Testing and OperationVon EverandCleanroom Technology: Fundamentals of Design, Testing and OperationNoch keine Bewertungen
- Production of Plasma Proteins for Therapeutic UseVon EverandProduction of Plasma Proteins for Therapeutic UseBewertung: 3 von 5 Sternen3/5 (5)
- Equipment Qualification in the Pharmaceutical IndustryVon EverandEquipment Qualification in the Pharmaceutical IndustryBewertung: 3.5 von 5 Sternen3.5/5 (3)
- Biocontamination Control for Pharmaceuticals and HealthcareVon EverandBiocontamination Control for Pharmaceuticals and HealthcareBewertung: 5 von 5 Sternen5/5 (1)
- Validation master plan Complete Self-Assessment GuideVon EverandValidation master plan Complete Self-Assessment GuideNoch keine Bewertungen
- Investigating SterilityTest FailuresDokument16 SeitenInvestigating SterilityTest FailuresMahesh_ChokshiNoch keine Bewertungen
- Basic Guide To Particle Counters and Particle CountigDokument60 SeitenBasic Guide To Particle Counters and Particle CountigJoel CunhaNoch keine Bewertungen
- Aseptic Processing Risk Assessment The Simplified Akers Agalloco MethodDokument55 SeitenAseptic Processing Risk Assessment The Simplified Akers Agalloco MethodDoan Chi ThienNoch keine Bewertungen
- The Manufacture of Sterile Pharmaceuticals and Liquid Medical Devices Using Blow-Fill-Seal Technology: Points to ConsiderVon EverandThe Manufacture of Sterile Pharmaceuticals and Liquid Medical Devices Using Blow-Fill-Seal Technology: Points to ConsiderNoch keine Bewertungen
- Validation Master Plan A Complete Guide - 2020 EditionVon EverandValidation Master Plan A Complete Guide - 2020 EditionNoch keine Bewertungen
- GMP in Pharmaceutical Industry: Global cGMP & Regulatory ExpectationsVon EverandGMP in Pharmaceutical Industry: Global cGMP & Regulatory ExpectationsBewertung: 5 von 5 Sternen5/5 (2)
- Regulatory Aspects of Pharmaceutical Quality System: Brief IntroductionVon EverandRegulatory Aspects of Pharmaceutical Quality System: Brief IntroductionNoch keine Bewertungen
- Software Validation A Complete Guide - 2020 EditionVon EverandSoftware Validation A Complete Guide - 2020 EditionNoch keine Bewertungen
- Qualification Procedure For Vial Washing Machine - Pharmaceutical GuidelinesDokument1 SeiteQualification Procedure For Vial Washing Machine - Pharmaceutical GuidelinesAli Goutas50% (2)
- Wfi UrsDokument43 SeitenWfi UrsMohsinShaikh100% (1)
- Good Manufacturing Practices (GMP) Modules for Pharmaceutical ProductsVon EverandGood Manufacturing Practices (GMP) Modules for Pharmaceutical ProductsNoch keine Bewertungen
- PQ WfiDokument9 SeitenPQ Wfisami2210Noch keine Bewertungen
- Visual Inspector Qualification PDFDokument7 SeitenVisual Inspector Qualification PDFrobin hasanNoch keine Bewertungen
- Pharmaceutical Quality by Design: A Practical ApproachVon EverandPharmaceutical Quality by Design: A Practical ApproachWalkiria S. SchlindweinNoch keine Bewertungen
- Autoclave Validation MSPDADokument35 SeitenAutoclave Validation MSPDAYessine Mrabet100% (1)
- Validation VialWasher OQ NIHDokument30 SeitenValidation VialWasher OQ NIHcongacon3aNoch keine Bewertungen
- Practical Approaches to Method Validation and Essential Instrument QualificationVon EverandPractical Approaches to Method Validation and Essential Instrument QualificationNoch keine Bewertungen
- Media Fill ChecklistDokument11 SeitenMedia Fill ChecklistSilke Igemann100% (1)
- Validation Master Plan A Complete Guide - 2021 EditionVon EverandValidation Master Plan A Complete Guide - 2021 EditionNoch keine Bewertungen
- Validation of Sterilization of AutoclavesDokument15 SeitenValidation of Sterilization of AutoclavesErich Hermann Günther Molina100% (3)
- Concepts of Quality Management in Pharmaceutical IndustryVon EverandConcepts of Quality Management in Pharmaceutical IndustryNoch keine Bewertungen
- How to Develop Robust Solid Oral Dosage Forms: From Conception to Post-ApprovalVon EverandHow to Develop Robust Solid Oral Dosage Forms: From Conception to Post-ApprovalNoch keine Bewertungen
- Validation Guide July2013Dokument37 SeitenValidation Guide July2013Herdiwan NovindraNoch keine Bewertungen
- Pharmaceutical Facilities: Design, Layouts and ValidationVon EverandPharmaceutical Facilities: Design, Layouts and ValidationBewertung: 4 von 5 Sternen4/5 (6)
- Cross Contamination Prevention in HVACDokument8 SeitenCross Contamination Prevention in HVAChuynhhaichauchauNoch keine Bewertungen
- Steam SterilizerDokument24 SeitenSteam Sterilizerj.k.kumar83% (6)
- How to Scale-Up a Wet Granulation End Point ScientificallyVon EverandHow to Scale-Up a Wet Granulation End Point ScientificallyBewertung: 4 von 5 Sternen4/5 (1)
- Pharmaceutical Industry Documents: 90 Pharmaceutical Quality Assurance Interview Questions & AnswersVon EverandPharmaceutical Industry Documents: 90 Pharmaceutical Quality Assurance Interview Questions & AnswersNoch keine Bewertungen
- Autoclave Validation ProtocolDokument23 SeitenAutoclave Validation ProtocolErum Manzoor100% (2)
- Prot OQ HVACDokument12 SeitenProt OQ HVACamrinNoch keine Bewertungen
- Accelerated Predictive Stability (APS): Fundamentals and Pharmaceutical Industry PracticesVon EverandAccelerated Predictive Stability (APS): Fundamentals and Pharmaceutical Industry PracticesFenghe QiuBewertung: 5 von 5 Sternen5/5 (1)
- Risk Management in Sterile EnvironmentsDokument30 SeitenRisk Management in Sterile EnvironmentsTim Sandle100% (4)
- Process ValidationDokument36 SeitenProcess ValidationRaghu Raj100% (1)
- Good Laboratory Practices and Compliance MonitoringVon EverandGood Laboratory Practices and Compliance MonitoringNoch keine Bewertungen
- Validation and Facility Design PDFDokument16 SeitenValidation and Facility Design PDFjpabloqfNoch keine Bewertungen
- Filter Validation Training-By PALLDokument85 SeitenFilter Validation Training-By PALLvipin_chaudhary100% (5)
- 30 ML Moulded Vial Filling OQDokument15 Seiten30 ML Moulded Vial Filling OQSubhash NaiduNoch keine Bewertungen
- Autoclave SVP Report 2016Dokument29 SeitenAutoclave SVP Report 2016Rajender Singh100% (1)
- How To Validate An AutoclaveDokument3 SeitenHow To Validate An AutoclaveqhpuongNoch keine Bewertungen
- Pharmaceutical Water - Pharmaceutical GuidelinesDokument6 SeitenPharmaceutical Water - Pharmaceutical GuidelinesAbou Tebba SamNoch keine Bewertungen
- Pantoprazole 40mg InjectionDokument36 SeitenPantoprazole 40mg Injectiondaizhussain004Noch keine Bewertungen
- Semi Solid Dosage Forms Manufacturing Tools Critical Process Parameters Strategies Optimization and ValidationDokument9 SeitenSemi Solid Dosage Forms Manufacturing Tools Critical Process Parameters Strategies Optimization and ValidationGeotamNoch keine Bewertungen
- PDA Technical Report 22 - SPADokument50 SeitenPDA Technical Report 22 - SPAErika100% (2)
- ISPE ArticleDokument12 SeitenISPE Articledrs_mdu48Noch keine Bewertungen
- An Overview of The Validation Approach For Moist Heat Sterilization, Part IIDokument9 SeitenAn Overview of The Validation Approach For Moist Heat Sterilization, Part IIqhpuongNoch keine Bewertungen
- How To Validate An AutoclaveDokument3 SeitenHow To Validate An AutoclaveqhpuongNoch keine Bewertungen
- Autoclave Mapping PDFDokument2 SeitenAutoclave Mapping PDFqhpuongNoch keine Bewertungen
- Operating Manual For S230 Conductivity MeterDokument49 SeitenOperating Manual For S230 Conductivity MeterqhpuongNoch keine Bewertungen
- AccuMedi - Endotoxin Indicator - Instruction For Use - DCV-2000Dokument2 SeitenAccuMedi - Endotoxin Indicator - Instruction For Use - DCV-2000qhpuongNoch keine Bewertungen
- Bảng dữ liệu hiệu chuẩn (260615)Dokument85 SeitenBảng dữ liệu hiệu chuẩn (260615)qhpuongNoch keine Bewertungen
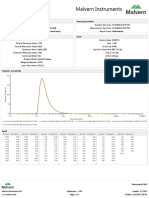
- Malvern Instruments Analysis: Measurement Details Measurement DetailsDokument1 SeiteMalvern Instruments Analysis: Measurement Details Measurement DetailsqhpuongNoch keine Bewertungen
- Thermo Hypersil GoldDokument20 SeitenThermo Hypersil GoldqhpuongNoch keine Bewertungen
- Valid Prcs S Gassy ST MsDokument6 SeitenValid Prcs S Gassy ST MsqhpuongNoch keine Bewertungen
- Manual Digi 28ssDokument7 SeitenManual Digi 28ssqhpuongNoch keine Bewertungen
- Phuong - DryheatDokument5 SeitenPhuong - DryheatqhpuongNoch keine Bewertungen
- BSBOPS601 Develop Implement Business Plans - SDokument91 SeitenBSBOPS601 Develop Implement Business Plans - SSudha BarahiNoch keine Bewertungen
- Tindara Addabbo, Edoardo Ales, Ylenia Curzi, Tommaso Fabbri, Olga Rymkevich, Iacopo Senatori - Performance Appraisal in Modern Employment Relations_ An Interdisciplinary Approach-Springer Internationa.pdfDokument278 SeitenTindara Addabbo, Edoardo Ales, Ylenia Curzi, Tommaso Fabbri, Olga Rymkevich, Iacopo Senatori - Performance Appraisal in Modern Employment Relations_ An Interdisciplinary Approach-Springer Internationa.pdfMario ChristopherNoch keine Bewertungen
- Practitioners Guide For Business Development Planning in FPOsDokument70 SeitenPractitioners Guide For Business Development Planning in FPOsMythreyi ChichulaNoch keine Bewertungen
- Terms and Conditions 27 06 PDFDokument4 SeitenTerms and Conditions 27 06 PDFShreyash NaikwadiNoch keine Bewertungen
- ATLAS HONDA Internship ReportDokument83 SeitenATLAS HONDA Internship ReportAhmed Aitsam93% (14)
- 6 AsianregionalismDokument32 Seiten6 AsianregionalismChandria Ford100% (1)
- Minor Ailments Services: A Starting Point For PharmacistsDokument49 SeitenMinor Ailments Services: A Starting Point For PharmacistsacvavNoch keine Bewertungen
- Ficha Tecnica 320D3 GCDokument12 SeitenFicha Tecnica 320D3 GCanahdezj88Noch keine Bewertungen
- Oxygen BarrierDokument20 SeitenOxygen BarrierKarina ArdizziNoch keine Bewertungen
- TQM BisleriDokument27 SeitenTQM BisleriDishank ShahNoch keine Bewertungen
- Bug Head - Fromjapanese To EnglishDokument20 SeitenBug Head - Fromjapanese To EnglishAnonymous lkkKgdNoch keine Bewertungen
- LT1256X1 - Revg - FB1300, FB1400 Series - EnglishDokument58 SeitenLT1256X1 - Revg - FB1300, FB1400 Series - EnglishRahma NaharinNoch keine Bewertungen
- Life Cycle Cost Analysis of Hvac System in Office ProjectsDokument3 SeitenLife Cycle Cost Analysis of Hvac System in Office ProjectsVashuka GhritlahreNoch keine Bewertungen
- Process Interactions PDFDokument1 SeiteProcess Interactions PDFXionNoch keine Bewertungen
- Gmo EssayDokument4 SeitenGmo Essayapi-270707439Noch keine Bewertungen
- FBW Manual-Jan 2012-Revised and Corrected CS2Dokument68 SeitenFBW Manual-Jan 2012-Revised and Corrected CS2Dinesh CandassamyNoch keine Bewertungen
- Quality in CRDokument10 SeitenQuality in CRkaushikcrNoch keine Bewertungen
- Majalah Remaja Islam Drise #09 by Majalah Drise - Issuu PDFDokument1 SeiteMajalah Remaja Islam Drise #09 by Majalah Drise - Issuu PDFBalqis Ar-Rubayyi' Binti HasanNoch keine Bewertungen
- Supreme Court of The United StatesDokument296 SeitenSupreme Court of The United StatesABC News PoliticsNoch keine Bewertungen
- Practical GAD (1-32) Roll No.20IF227Dokument97 SeitenPractical GAD (1-32) Roll No.20IF22720IF135 Anant PatilNoch keine Bewertungen
- Kompetensi Sumber Daya Manusia SDM Dalam Meningkatkan Kinerja Tenaga Kependidika PDFDokument13 SeitenKompetensi Sumber Daya Manusia SDM Dalam Meningkatkan Kinerja Tenaga Kependidika PDFEka IdrisNoch keine Bewertungen
- Tendernotice 1Dokument42 SeitenTendernotice 1Hanu MittalNoch keine Bewertungen
- Heat Exchanger Designing Using Aspen PlusDokument6 SeitenHeat Exchanger Designing Using Aspen PlusMeethiPotterNoch keine Bewertungen
- NX CAD CAM AutomationDokument12 SeitenNX CAD CAM AutomationfalexgcNoch keine Bewertungen
- Rehabilitation and Retrofitting of Structurs Question PapersDokument4 SeitenRehabilitation and Retrofitting of Structurs Question PapersYaswanthGorantlaNoch keine Bewertungen
- Dr. Najeebuddin Ahmed: 969 Canterbury Road, Lakemba, Sydney, NSW, Australia, 2195Dokument2 SeitenDr. Najeebuddin Ahmed: 969 Canterbury Road, Lakemba, Sydney, NSW, Australia, 2195Najeebuddin AhmedNoch keine Bewertungen
- Project Management: Chapter-2Dokument26 SeitenProject Management: Chapter-2Juned BhavayaNoch keine Bewertungen
- Unit 5 Andhra Pradesh.Dokument18 SeitenUnit 5 Andhra Pradesh.Charu ModiNoch keine Bewertungen
- Purchases + Carriage Inwards + Other Expenses Incurred On Purchase of Materials - Closing Inventory of MaterialsDokument4 SeitenPurchases + Carriage Inwards + Other Expenses Incurred On Purchase of Materials - Closing Inventory of MaterialsSiva SankariNoch keine Bewertungen
- Ministry of Education Musala SCHDokument5 SeitenMinistry of Education Musala SCHlaonimosesNoch keine Bewertungen