Das könnte Ihnen auch gefallen
- ProblemasredescristalinasDokument8 SeitenProblemasredescristalinasPEDROLOERANoch keine Bewertungen
- Proyección estereográfica en ingeniería geológicaDokument10 SeitenProyección estereográfica en ingeniería geológicaMartin VarelaNoch keine Bewertungen
- Sesión Ix-X CMCM 2018Dokument30 SeitenSesión Ix-X CMCM 2018alexNoch keine Bewertungen
- El proceso SART: Una opción rentable para el tratamiento de minerales auríferos con cobreDokument18 SeitenEl proceso SART: Una opción rentable para el tratamiento de minerales auríferos con cobreMoe HoNoch keine Bewertungen
- Construcción y Analisis de Un VariogramaDokument15 SeitenConstrucción y Analisis de Un VariogramaConstanza Caro RiquelmeNoch keine Bewertungen
- Clase 3-Ejercicios de Imperfecciones CristalinasDokument7 SeitenClase 3-Ejercicios de Imperfecciones Cristalinasjosselynra0% (3)
- Flotación Cu-MoDokument10 SeitenFlotación Cu-MoAldentaraNoch keine Bewertungen
- Ley de corte minería abiertaDokument104 SeitenLey de corte minería abiertaOliver Ventocilla PadillaNoch keine Bewertungen
- Mineralogia Sistematica IDokument79 SeitenMineralogia Sistematica IJoan Irvin100% (2)
- Análisis Termodinámico de La Lixiviación de Minerales Sulfurados de CobreDokument4 SeitenAnálisis Termodinámico de La Lixiviación de Minerales Sulfurados de CobreGrace Torres SarmientoNoch keine Bewertungen
- GeometalurgiaDokument19 SeitenGeometalurgiaManuel Guerreros MezaNoch keine Bewertungen
- Economía Minera ApuntesDokument24 SeitenEconomía Minera ApuntesGerardo Andres González DiazNoch keine Bewertungen
- Optimización de la ley de corte para maximizar el VPN en minería a cielo abiertoDokument8 SeitenOptimización de la ley de corte para maximizar el VPN en minería a cielo abiertoJhoan Crhistian HCNoch keine Bewertungen
- Fórmulas Empíricas PDFDokument4 SeitenFórmulas Empíricas PDFEduardo Vera SanhuezaNoch keine Bewertungen
- Fusion de Concentrado-Tesis Rodolfo BerriosDokument104 SeitenFusion de Concentrado-Tesis Rodolfo BerriosRDario Dario Rosemary100% (1)
- Conc. Ii - 14 - 2 - Balance MetalúrgicoDokument41 SeitenConc. Ii - 14 - 2 - Balance MetalúrgicoAlex Espinoza AlvaradoNoch keine Bewertungen
- PLAN-FINAL-ELIANA-Y-DEYMI TesisDokument23 SeitenPLAN-FINAL-ELIANA-Y-DEYMI TesisEliana Ivette Castrejon Caruanambo100% (1)
- Examen Comercializacion de Minerales y Metales 2012Dokument3 SeitenExamen Comercializacion de Minerales y Metales 2012Frank AguilarNoch keine Bewertungen
- Ley de Corte y Balance FinalDokument23 SeitenLey de Corte y Balance FinalKevin Rojas AtalayaNoch keine Bewertungen
- Ley EquivalenteDokument4 SeitenLey EquivalentepabloNoch keine Bewertungen
- KrigeajeDokument15 SeitenKrigeajeHAROLDNoch keine Bewertungen
- Minería superficial: cálculos de ley de corte y diseño de pitDokument15 SeitenMinería superficial: cálculos de ley de corte y diseño de pitJesus Condori HuarillocllaNoch keine Bewertungen
- Flotación de minerales: proceso y agentesDokument20 SeitenFlotación de minerales: proceso y agentesJuan Manuel Avalos RodriguezNoch keine Bewertungen
- PDF Informe Quincenal Mineria Como Se Calcula El Valor de Los Concentrados de MineralesDokument6 SeitenPDF Informe Quincenal Mineria Como Se Calcula El Valor de Los Concentrados de MineraleslucianotigreNoch keine Bewertungen
- Ley de Corte-Plan Mens. Tajo Abie..Dokument21 SeitenLey de Corte-Plan Mens. Tajo Abie..Caleb Adrian Valenzuela SalvatierraNoch keine Bewertungen
- Muestreo MineroDokument43 SeitenMuestreo Minerosantiago rhNoch keine Bewertungen
- Transporte de concentrados minerales: sistemas y requerimientosDokument18 SeitenTransporte de concentrados minerales: sistemas y requerimientosLuigi Garcia Cueva100% (1)
- Copia de Cuestionario de Aglomerado-2Dokument6 SeitenCopia de Cuestionario de Aglomerado-2Florencia Gallardo100% (1)
- Teoria Del SoporteDokument28 SeitenTeoria Del SoporteARTURO PICON MONTOYANoch keine Bewertungen
- Fileteado, Festoneado y Biselado de EsquinasDokument2 SeitenFileteado, Festoneado y Biselado de EsquinasDeya NiraNoch keine Bewertungen
- G2-ACSM-Flujo de Caja LibreDokument51 SeitenG2-ACSM-Flujo de Caja LibreLuis-Fernando Haro JaraNoch keine Bewertungen
- Clase 1 - Introduccion y Fundamentos de La Comercializacion de Minerales PDFDokument33 SeitenClase 1 - Introduccion y Fundamentos de La Comercializacion de Minerales PDFVania Malca NeyraNoch keine Bewertungen
- Catálogos de Celdas de FlotaciónDokument3 SeitenCatálogos de Celdas de FlotaciónPaulaNoch keine Bewertungen
- Tarea DecimoDokument7 SeitenTarea DecimoKaren YaimaNoch keine Bewertungen
- Segunda Práctica de Comercialización de MineralesDokument39 SeitenSegunda Práctica de Comercialización de MineralesChristian Alexis Rosas PelaezNoch keine Bewertungen
- Ejemplo - Estación DCR PDFDokument1 SeiteEjemplo - Estación DCR PDFTJ NolascoNoch keine Bewertungen
- Evaluacion de Proyectos MienrosDokument16 SeitenEvaluacion de Proyectos MienrosmisaelNoch keine Bewertungen
- Ley de Corte 1Dokument35 SeitenLey de Corte 1RodrigoAlexisTapiaAguirreNoch keine Bewertungen
- ProblemasComercializacionMineralesDokument7 SeitenProblemasComercializacionMineralesbryanNoch keine Bewertungen
- El Estimador de Costos oDokument7 SeitenEl Estimador de Costos oCarlos Lei BrioNoch keine Bewertungen
- Parcial de Estructura y Propiedades FIGMMDokument5 SeitenParcial de Estructura y Propiedades FIGMMEd LCNoch keine Bewertungen
- Proceso SARTDokument4 SeitenProceso SARTAlexis YañezNoch keine Bewertungen
- Mrozek, S. Et Al. (2020)Dokument13 SeitenMrozek, S. Et Al. (2020)OLIVER100% (1)
- Criterios de Diseño de Tuberias CiegasDokument10 SeitenCriterios de Diseño de Tuberias CiegasRommel ValladaresNoch keine Bewertungen
- El Código JorcDokument37 SeitenEl Código JorcFer CremaNoch keine Bewertungen
- Modelos de Difusión AtmosféricaDokument5 SeitenModelos de Difusión AtmosféricaDiana De Los Santos MendozaNoch keine Bewertungen
- Recuperación Por Aglomeración de Carbón y Petróleo-Ecuacion de YoungDokument23 SeitenRecuperación Por Aglomeración de Carbón y Petróleo-Ecuacion de Youngvictor cuadrosNoch keine Bewertungen
- Informe de prácticas pre-profesionales en planta de beneficio de mineralesDokument61 SeitenInforme de prácticas pre-profesionales en planta de beneficio de mineralesFRANCOFIGMM100% (1)
- Comercializacion de La PlataDokument24 SeitenComercializacion de La PlataOmar Flores CapchaNoch keine Bewertungen
- 02 Recursos RecuperablesDokument147 Seiten02 Recursos RecuperablesNancy SalcedoNoch keine Bewertungen
- Tema09 MG Leymineral Valoreconmico 141108144150 Conversion Gate02Dokument34 SeitenTema09 MG Leymineral Valoreconmico 141108144150 Conversion Gate02izamielccNoch keine Bewertungen
- Metodo de Transformacion de Triangulo A EstrellaDokument27 SeitenMetodo de Transformacion de Triangulo A EstrellaLeonardo HornaNoch keine Bewertungen
- Balance Metalúrgico CemrobDokument12 SeitenBalance Metalúrgico CemrobNilton QuispeNoch keine Bewertungen
- Estructura CristalinaDokument85 SeitenEstructura Cristalinalisbeth camachoNoch keine Bewertungen
- CLASE CristalografiaDokument49 SeitenCLASE CristalografiaJesus Noel Mendoza VenturaNoch keine Bewertungen
- Estructura de Los Sólidos 2012-1Dokument38 SeitenEstructura de Los Sólidos 2012-1Marlon TeveNoch keine Bewertungen
- Estructuras cristalinas en ingeniería civilDokument34 SeitenEstructuras cristalinas en ingeniería civilIsabel Jaen MurgasNoch keine Bewertungen
- Tema 3 Estructura Cristalina PDFDokument49 SeitenTema 3 Estructura Cristalina PDFYanela Katherine Alva DiazNoch keine Bewertungen
- 2-Estructura de Los Solidos CristalinosDokument19 Seiten2-Estructura de Los Solidos CristalinosJeferson CordovaNoch keine Bewertungen
- Estructura de Los Metales PDFDokument21 SeitenEstructura de Los Metales PDFMaxidvpNoch keine Bewertungen
- La Revolución Mexicana y La Importancia de La ConstituciónDokument10 SeitenLa Revolución Mexicana y La Importancia de La ConstituciónClaudia KarlaNoch keine Bewertungen
- Ing. de Aleac. Ornamental, MapaDokument15 SeitenIng. de Aleac. Ornamental, MapaClaudia KarlaNoch keine Bewertungen
- ramanEXPO CERAMICOS2Dokument30 SeitenramanEXPO CERAMICOS2Claudia KarlaNoch keine Bewertungen
- Grafico Del Nivel de La Tasa de Referencia BANXICODokument2 SeitenGrafico Del Nivel de La Tasa de Referencia BANXICOClaudia KarlaNoch keine Bewertungen
- CursosDokument8 SeitenCursosClaudia Karla33% (3)
- TABLASDokument18 SeitenTABLASClaudia KarlaNoch keine Bewertungen
- Soldadura de VentaneriaDokument47 SeitenSoldadura de VentaneriaClaudia KarlaNoch keine Bewertungen
- Ing. de Aleac. Ornamental, MapaDokument15 SeitenIng. de Aleac. Ornamental, MapaClaudia KarlaNoch keine Bewertungen
- Nivel Básico 1 DCDokument160 SeitenNivel Básico 1 DCClaudia KarlaNoch keine Bewertungen
- ÍndiceDokument7 SeitenÍndiceClaudia KarlaNoch keine Bewertungen
- SUSTENTABILIDADDokument5 SeitenSUSTENTABILIDADClaudia KarlaNoch keine Bewertungen
- Ciclo Del Carbono y Del OxigenoDokument51 SeitenCiclo Del Carbono y Del OxigenoClaudia KarlaNoch keine Bewertungen
- Tareahidrockhm 3Dokument2 SeitenTareahidrockhm 3Claudia KarlaNoch keine Bewertungen
- Bancos y Otros Elementos de Descanso para El Uso PúblicoDokument15 SeitenBancos y Otros Elementos de Descanso para El Uso PúblicoClaudia KarlaNoch keine Bewertungen
- Poster EspadaDokument1 SeitePoster EspadaClaudia KarlaNoch keine Bewertungen
- Web Informe GM BMV 2015 PDFDokument225 SeitenWeb Informe GM BMV 2015 PDFClaudia KarlaNoch keine Bewertungen
- Triángulos en el arteDokument7 SeitenTriángulos en el arteClaudia KarlaNoch keine Bewertungen
- Claudia MQE Tornillo1Dokument18 SeitenClaudia MQE Tornillo1Claudia KarlaNoch keine Bewertungen
- CuentosDokument8 SeitenCuentosClaudia KarlaNoch keine Bewertungen
- HIERRODokument49 SeitenHIERROClaudia Karla100% (1)
- EnsayooooDokument8 SeitenEnsayooooClaudia KarlaNoch keine Bewertungen
- Formato Del Protocolo de Ejercicios de LaboratorioDokument2 SeitenFormato Del Protocolo de Ejercicios de LaboratorioClaudia KarlaNoch keine Bewertungen
- 7 Hábitos de La Gente Altamente EfectivaDokument10 Seiten7 Hábitos de La Gente Altamente EfectivaFelipe LoveraNoch keine Bewertungen
- Transferencia de Calor Por Convección Forzada CKHMDokument4 SeitenTransferencia de Calor Por Convección Forzada CKHMClaudia KarlaNoch keine Bewertungen
- CKHM Lab QO1 JuevesDokument7 SeitenCKHM Lab QO1 JuevesClaudia KarlaNoch keine Bewertungen
- Fpe MemoriaDokument6 SeitenFpe MemoriaClaudia KarlaNoch keine Bewertungen
- Guía para obtener patentes y modelos de utilidadDokument106 SeitenGuía para obtener patentes y modelos de utilidadDaniel OrtegaNoch keine Bewertungen
- Normas y requisitos para exentar seminario de fundición UNAMDokument2 SeitenNormas y requisitos para exentar seminario de fundición UNAMClaudia KarlaNoch keine Bewertungen
- Ing Economica Tecate y At&tDokument2 SeitenIng Economica Tecate y At&tClaudia KarlaNoch keine Bewertungen
- Ejercicios Resueltos.Dokument15 SeitenEjercicios Resueltos.checho00% (2)
- Redes de BravaisDokument3 SeitenRedes de BravaisNicolas SalazarNoch keine Bewertungen
- S20 - Química - 4º Secundaria - EvaluaciónDokument3 SeitenS20 - Química - 4º Secundaria - EvaluaciónalanNoch keine Bewertungen
- Hds Esmalte EpoxicoDokument4 SeitenHds Esmalte EpoxicoDaniela Santibañez0% (1)
- Dilatación volumétrica: concepto, fórmulas y ejemplosDokument4 SeitenDilatación volumétrica: concepto, fórmulas y ejemplosAnaLuciaNoch keine Bewertungen
- Formato de Entrega Tarea 2Dokument12 SeitenFormato de Entrega Tarea 2liliana burbanoNoch keine Bewertungen
- Coeficiente de DilataciónDokument3 SeitenCoeficiente de DilataciónIshaell Amores100% (1)
- Calcantita MineralogiaDokument7 SeitenCalcantita MineralogiaMaydde Cutipa0% (1)
- Trabajo de CristalograficaDokument9 SeitenTrabajo de CristalograficaWilliamsRafaelMataRimacNoch keine Bewertungen
- Atlas Copco 4.11 TraduccionDokument122 SeitenAtlas Copco 4.11 Traduccioncristian saldivar100% (2)
- Presentacion PinturasDokument42 SeitenPresentacion Pinturasoscar alarconNoch keine Bewertungen
- Curso ChancadoDokument162 SeitenCurso ChancadoALDO ROGERNoch keine Bewertungen
- Influencia Del Tiempo y La Temperatura en La Velocidad de Corrosión Del Acero Aisi 316 en Fase Gaseosa en El Procesamiento de Un Crudo PesadoDokument56 SeitenInfluencia Del Tiempo y La Temperatura en La Velocidad de Corrosión Del Acero Aisi 316 en Fase Gaseosa en El Procesamiento de Un Crudo PesadoAndres YayaNoch keine Bewertungen
- Sem 14 Tarea Practica 2022-IDokument13 SeitenSem 14 Tarea Practica 2022-Imaria juanaNoch keine Bewertungen
- Introducción a la elaboración de productos de aseo y cosméticos - Materiales necesariosDokument13 SeitenIntroducción a la elaboración de productos de aseo y cosméticos - Materiales necesariosmilovillaNoch keine Bewertungen
- CARACTERISTICAS TECNICAS MINIMAS EquiposDokument6 SeitenCARACTERISTICAS TECNICAS MINIMAS EquiposYedira BecerraNoch keine Bewertungen
- Examen ICFES Química OrgánicaDokument14 SeitenExamen ICFES Química OrgánicaHamilton A. MartinezNoch keine Bewertungen
- Ficha Tecnica AZ 20 - R-410ADokument2 SeitenFicha Tecnica AZ 20 - R-410ATato MayoNoch keine Bewertungen
- Artículo Hidroxiapatita.Dokument7 SeitenArtículo Hidroxiapatita.SantiagoCanoMolinaNoch keine Bewertungen
- Flujo Monofásico en Sistemas Homogéneos Lineales y RadialesDokument8 SeitenFlujo Monofásico en Sistemas Homogéneos Lineales y RadialesAlessandro GarciaNoch keine Bewertungen
- SEMINARIO 3 Cinética Química, Equilibrio Químico y Equilibrio Acido Base 123 ADokument12 SeitenSEMINARIO 3 Cinética Química, Equilibrio Químico y Equilibrio Acido Base 123 ASandra CordovaNoch keine Bewertungen
- Acuifero MorroaDokument20 SeitenAcuifero MorroaJavier ReyesNoch keine Bewertungen
- Práctica 1. Preparación de Disoluciones y Determinación de La Concentración de Una Disolución Por Medio de Una Valoración (Titulación)Dokument8 SeitenPráctica 1. Preparación de Disoluciones y Determinación de La Concentración de Una Disolución Por Medio de Una Valoración (Titulación)jesusNoch keine Bewertungen
- La ElectroquìmicaDokument8 SeitenLa ElectroquìmicaAnonymous iWVC2RKzSNoch keine Bewertungen
- Proyecto Metalicas - Casi ListoDokument44 SeitenProyecto Metalicas - Casi ListoRyu Rey RogerNoch keine Bewertungen
- Determinación de la velocidad enzimática de la fosfatasa ácida de bovinoDokument17 SeitenDeterminación de la velocidad enzimática de la fosfatasa ácida de bovinoIgnacio ArFuNoch keine Bewertungen
- Anilina LiquidaDokument2 SeitenAnilina LiquidaCamilo FernandezNoch keine Bewertungen
- Determinación de acidez en jugo comercial mediante valoración ácido-baseDokument4 SeitenDeterminación de acidez en jugo comercial mediante valoración ácido-baseAlexa BarragánNoch keine Bewertungen
- Natal Branco PARTITURA 3Dokument4 SeitenNatal Branco PARTITURA 3Cadu BarcelosNoch keine Bewertungen
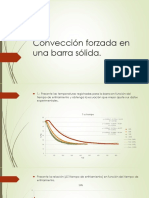
- Convección forzada en barra sólida: coeficiente de transferencia de energíaDokument12 SeitenConvección forzada en barra sólida: coeficiente de transferencia de energíaKarliita LomNoch keine Bewertungen