Beruflich Dokumente
Kultur Dokumente
Pervasive Contaminations in Sequencing Experiments Are A Major Source of False Genetic Variability: A Mycobacterium Tuberculosis Meta-Analysis
Hochgeladen von
kjhgfghjkOriginaltitel
Copyright
Verfügbare Formate
Dieses Dokument teilen
Dokument teilen oder einbetten
Stufen Sie dieses Dokument als nützlich ein?
Sind diese Inhalte unangemessen?
Dieses Dokument meldenCopyright:
Verfügbare Formate
Pervasive Contaminations in Sequencing Experiments Are A Major Source of False Genetic Variability: A Mycobacterium Tuberculosis Meta-Analysis
Hochgeladen von
kjhgfghjkCopyright:
Verfügbare Formate
bioRxiv preprint first posted online Aug. 29, 2018; doi: http://dx.doi.org/10.1101/403824.
The copyright holder for this preprint
(which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity.
It is made available under a CC-BY-NC 4.0 International license.
Pervasive contaminations in sequencing
experiments are a major source of false genetic
variability: a Mycobacterium tuberculosis
meta-analysis
Galo A. Goig1 , Silvia Blanco2 , Alberto L. Garcia-Basteiro2,3,4 , and Iñaki Comas1,5 *
1
Institute of Biomedicine of Valencia, IBV-CSIC, Valencia, Spain
2
Centro de Investigaçao em Saúde de Manhiça (CISM), Maputo, Moçambique
3
Global Health Institute Barcelona (ISGlobal), Barcelona, Spain
4
Amsterdam Institute for Global Health and Development, Amsterdam University Medical Centers, Amsterdam, Netherlands
5
CIBER in Epidemiology and Public Health, Madrid, Spain
Whole genome sequencing (WGS) is now playing a central role hindering high-throughput sequencing data interpretation are
in the control and study of tuberculosis. Factors hindering ge- usually overlooked.
nomic data interpretation can difficult our understanding of the Over the last years, groups working in different areas have
pathogen biology and lead to incorrect clinical predictions. An-
shown that DNA contaminations is one factor potentially
alyzing an extensive Mycobacterium tuberculosis dataset com-
prising more than 1,500 sequencing samples from different pub-
misleading biological conclusions (7–9). However, most bac-
lished works, with additional samples sequenced in our labora- terial genomic studies are still neglecting the impact of poten-
tory, we find that contamination with non-target DNA is a com- tial contaminations with non-target DNA in their analyses.
mon phenomenon among WGS studies. By using this data and This stems from the general assumption that pure culture se-
in-silico simulations, we show that even subtle contaminations quencings only yield reads from the target organism and, if
can produce dozens of false variants and large miscalculations any, contaminating reads can hardly map to the target ref-
of allele frequencies, often leading to errors that are very hard to erence genome. As the decentralization of high-throughput
detect and propagate through the analysis. In our dataset, 94% sequencing and its associated analysis allow more groups to
of the polymorphic positions were incorrectly identified due to conduct WGS studies, controlling the factors that compro-
contaminations. We exemplify the consequences of these errors mise the outcomes accuracy becomes crucial. This can be
in the context of clinical predictions for all the studies analyzed,
particularly relevant when studying bacterial organisms with
and demonstrate that unexpected contaminations suppose a ma-
jor pitfall in WGS studies. In addition, we present an approach
low genetic diversity, for which evolutionary inferences and
based on the removal of contaminant reads that shows an out- diagnostic may depend on few genetic variants.
standing performance analyzing both clean and contaminated Mycobacterium tuberculosis (MTB) represents a perfect ex-
data. Based in our findings, applicable to most of organisms, ample of such an organism. Tuberculosis is now recognized
we urge for the implementation of contamination-aware analy- as the deadliest infectious disease in the world and one quar-
sis pipelines. ter of the global population is estimated to be infected (10).
Tuberculosis | Whole-genome sequencing | Contamination | Variant calling Due to its long culture-based diagnostic turnarounds, WGS
Correspondence: icomas@ibv.csic.es is being extensively used not only to study its biology, but
also in the clinical context(11, 12). Particularly, the ability of
sequencing the complete genome of the pathogen from com-
Introduction plex clinical samples will suppose a major breakthrough, now
Whole genome sequencing (WGS) has enhanced the study pursued by several groups (13, 14).
of complex biological phenomena in bacteria, such as pop- Here we use an extensive dataset comprising more than 1500
ulation dynamics, host adaptation or outbreaks of micro- MTB sequencing samples coming from different WGS stud-
bial infections(1, 2). Democratization of high-throughput se- ies along with in-silico simulated data to assess the impact
quencing technologies and continuous improvements in lab- of contamination in WGS analysis. We show that presence
oratory procedures are also turning WGS into a promising of non-target reads is a common phenomenon among WGS
alternative for the clinical diagnosis and surveillance of sev- studies, even when sequencing is performed from pure cul-
eral pathogenic species (3–6). Since most genomic studies ture isolates. Importantly, we demonstrate that subtle con-
are based on the identification of nucleotide polymorphisms taminations represent a major pitfall, since they are prevalent,
and calculation of allele frequencies, the accuracy of the vari- hard to detect, and can introduce dozens of false variants.
ant calling process is pivotal to draw biological conclusions In our dataset, a great proportion of the estimated genetic
from WGS data. Notwithstanding, the performance of the variability was due to contamination, and allele frequency
analysis pipelines is seldomly evaluated, and possible factors calculations, the foundation of most genomic studies, were
Goig et al. | bioRχiv | August 29, 2018 | 1–12
bioRxiv preprint first posted online Aug. 29, 2018; doi: http://dx.doi.org/10.1101/403824. The copyright holder for this preprint
(which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity.
It is made available under a CC-BY-NC 4.0 International license.
largely altered. These alterations can lead to misinterpreta- tory microbiota, clinically common NTM and human. As
tion of WGS data as we demonstrate for two relevant appli- expected, conserved genes like the 16S, rpoB or the tR-
cations: epidemiology and drug resistance prediction. In ad- NAs, constitute hotspots where contaminant sequences are
dition, we provide a contamination-aware workflow based on easily aligned to. However, non-MTB alignments are pro-
a taxonomic filter that exhibits an outstanding performance, duced across the reference genome (Figure 2a). Naturally,
allowing a robust analysis even for highly contaminated sam- this is dependent on the phylogenetic relationship of the con-
ples, such as sequencings from complex clinical specimens. taminant organism to the one being studied. For example,
many more sequencing reads from Nocardia are aligned to
Results the Mycobacterium tuberculosis complex (MTBC) reference
genome than from distantly related organisms such as Strep-
DNA contamination is pervasive across WGS MTB tococcus . Non-tuberculosis mycobacteria represent the best
studies and sample types. To look for contaminant DNA example of this as their read mappings can produce high se-
in WGS datasets, we used Kraken (15) to taxonomically quencing depths along the MTBC reference genome. Impor-
classify the sequencing reads of 1553 sequencing runs from tantly, human reads, which are usually the major concern in
eight different MTB studies (here referred as the experimen- clinical samples, did not produced alignments at all.
tal dataset, see Methods). According to Kraken classifica-
tions, contamination is present to some extent in all stud- Next, we assessed the performance of two contamination-
ies analyzed (Figure 1). As expected, sequencings from di- aware approaches to avoid errors introduced by non-target
rect clinical samples and early positive mycobacterial growth reads: a filter removing low quality alignments, and a taxo-
indicator tubes (MGIT), which are inoculated with primary nomic filter removing non-MTBC reads. By measuring the
clinical samples, present higher levels of contamination in mean sequencing depth obtained along the genome in 100-
terms of both the number of samples contaminated and the bp windows, we observed that both methods greatly reduced
proportion of non-MTB DNA within them. Common con- the number of non-MTB mappings (see Additional file 1).
taminants for these samples comprise human DNA and bac- Remarkably, whereas the mapping filter was unable to elim-
teria usually found in oral and respiratory cavities like Pseu- inate contaminant alignments in several conserved regions,
domonas, Rothia, Streptococcus or Actinomyces (16, 17), and specially the 16S gene, the taxonomic filter displayed an
can constitute virtually all reads in some samples. Impor- outstanding performance eliminating all non-MTB mappings
tantly, contamination was also detected in studies in which with the only exception of some M. avium reads (Additional
the DNA sequenced came from pure culture isolates. For file 2).
instance, Bacillus, Negativicoccus and Enterococcus repre- In order to evaluate how these contaminant alignments can
sented up to the 68%, 58% and 32% respectively of different affect WGS analysis, we simulated mock samples where se-
samples from the KwaZulu study. Strikingly, 17 out of 73 quencing reads from the reference genome were mixed with
samples from Nigeria were identified as Staphylococcus au- different proportions of non-MTBC organisms. In addition,
reus (92% to 99% of reads), probably due to a mistake during we generated a version of the reference genome where ran-
data uploading or mislabeling. The deep sequencing dataset dom mutations were introduced. This allowed us to quan-
was mostly free of contamination, with the exception of two tify both false positive and negative SNPs (see Methods). As
samples for which a 3.32% of Acinetobacter baumannii and shown in Figure 2b, slight contaminations can be responsi-
a 2.83% of non-tuberculosis mycobacteria (NTM) was iden- ble for a high number of false positive and negative variant
tified (representing 795,887 and 920,379 reads respectively). calls. Predictably, the vast majority of the false positives cor-
Remarkably, Kraken left a high proportion of reads unclas- responded to variable SNPs (vSNPs) and false positive fixed
sified in many instances. This could be mainly due to ei- SNPs (fSNPs) only originated from extreme contaminations.
ther the absence of the organism from the database, or hard Reduction of non-MTBC alignments mediated by the map-
to classify sequences. Indeed, when using the NCBI blastn ping filter led to a corresponding reduction in the number of
(18) to search a random subset of unclassified reads in the false positive SNPs, although many erroneous variants still
non-redundant database (nr), we observed three main pat- being called. In some cases, the number of false negatives
terns. Reads that either did not produce significant matches was higher denoting the elimination of correct MTBC align-
with any organism, or came from eukaryotes not present in ments by this filter. By contrast, the taxonomic filter elim-
our Kraken database; reads that produced partial alignments inated nearly all erroneous SNP calls. Its performance was
with many different taxa; and reads that produced good align- only compromised by contamination with Mycobacterium
ments, even with Mycobacterium tuberculosis, but having avium. In this case, the taxonomic filter was not able to avoid
alignment identities around 90%, what makes Kraken unable all false positive and negative SNPs introduced by contami-
to find exact matches of 31 base pairs. nant M. avium reads. For example, when a 5% of M. avium
was present, the 4,353 false positive SNPs and 71 false neg-
Evaluating the impact of contamination in WGS anal- ative SNPs identified with a conventional pipeline, were re-
ysis with simulated data. Firstly, we evaluated how non- duced to 41 and 4 respectively applying the taxonomic filter.
MTB sequencing reads map to the MTB reference genome. Importantly, applying the taxonomic filter to the uncontami-
In order to do this, we performed alignments of simulated nated mock sample of the reference genome did not produce
sequencings for 45 organisms, including oral and respira- any false negative.
2 | bioRχiv Goig et al. | Impact of contaminations in WGS
bioRxiv preprint first posted online Aug. 29, 2018; doi: http://dx.doi.org/10.1101/403824. The copyright holder for this preprint
(which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity.
It is made available under a CC-BY-NC 4.0 International license.
Since many of the contaminant alignments are produced in focused our analysis on the subtle changes produced by mod-
conserved genes, all these erroneous calls led to the pre- erate to light contaminations. To do this, we analyzed only
diction of multiple false antibiotic resistances when using a the samples that were within the range of 650-1400 fSNPs
conventional pipeline (see Additional file 1). Importantly, to the MTBC ancestral genome when using a conventional
many of these false predictions corresponded to first line pipeline, and had at least the 50% of sequencing reads clas-
drugs, classified as high confidence mutations according to sified as MTBC (n=1381 samples; 89.55% of the experimen-
the PhyResSE reference catalog(19) (Additional file 3). tal dataset). These represent the samples in which neither
the contaminations nor the errors produced would be easily
Normalization of SNPs by mapping to an MTBC an- detected. Comparing with the taxonomic filter approach re-
cestral genome. Normalizing the number of SNPs between vealed that variation in the number of SNPs (vSNPs + fSNPs)
samples allowed the direct comparison of WGS outcomes is a common fact among studies, with no exception (Figure
between studies and a detailed analysis of the impact pro- 4). Globally, around the 10% of samples differed in at least
duced by subtle contaminations. This normalization was pos- 10 SNPs. In samples with 50% to 90% of MTBC (n=63), the
sible by mapping to an inferred MTBC ancestral genome median change in the number of SNPs was 81 (IQR=186).
(20). To illustrate this, we compared the outcomes of a stan- Notably, these differences are observed in samples with slight
dard pipeline in the experimental dataset using the afore- contaminations as well, as in 66 samples with 90% to 97% of
mentioned ancestral genome and the conventional reference MTBC we observed a median change of 13 SNPs (IQR=51).
strain for MTB, H37Rv. In order to avoid noise introduced In fact, as shown in Figure 4, dozens of false SNPs can be
by possible contaminations, we only analyzed the samples called even for samples that could be considered “pure”.
with more than the 99% of the reads classified as MTBC and Analyzing samples with a percentage of MTBC reads greater
producing at least 40X of median sequencing depth (n=983). than the 99% (n=1009; 72%) allowed us to confirm, along
As shown in Figure 3, when mapping to the inferred MTBC with the simulated data, that the taxonomic filter itself did
ancestor (Figure 3.a), the number of fSNPs for all samples not impact the results presented above. In these samples, the
stabilized around a very narrow range, in opposite to the re- median difference in SNPs was one (IQR=2) before and after
sults obtained when mapping to H37Rv (Figure 3.b). In fact, eliminating contamination. Indeed, for 377 of these samples
99% of samples coming from human MTB strains (n=964) (27%) no change in SNPs was observed at all.
were within a range of 668-953 fSNPs (median=860 fSNPs,
interquartile range (IQR)=87). In contrast, when mapping Variant frequencies are extremely sensitive to contam-
to the reference strain, this range is dramatically extended to inant sequences. Due to the relevance of variable posi-
381-1802 fSNPs (median=794 fSNPs, IQR=503). In the lat- tions and their individual frequencies in genome-based stud-
ter case, the lineage 4.10 strains, which H37Rv belongs to, ies, we compared all the vSNPs called for the conventional
are much closer (408 fSNPs on average) than lineage 2 (1178 and the taxonomic filter workflows. A total of 269,938 non-
fSNPs), or lineage 1 (1769 fSNPs) strains. When mapping to redundant variable positions were predicted by the conven-
the MTBC ancestral genome, the remaining 1% of samples tional analysis pipeline. When applying the taxonomic fil-
were out of this range due to low sequencing depths, coinfec- ter, this number was dramatically reduced to only 16,464
tions where many fSNP are lost due to mixed calls, and high non-redundant variable positions and, consequently, the 94%
contaminations as will be detailed in the following sections. of the variants detected were attributed to noise introduced
Animal strains had also a higher number of fSNPs to the an- by contaminants. Importantly, non-target reads produced
cestral reference than human strains (895-1325 fSNPs, me- large fluctuations in SNP frequencies. The median differ-
dian=936 fSNPs, IQR=248, n=19), what is probably linked ence in frequency for all vSNPs was 26% (IQR=39%) in
to longer branches in the phylogeny. Based on this analysis contaminated samples (MTBC < 99%; n=383; 28% of sam-
we set up an expected range of SNPs for any given MTB sam- ples), and the magnitude of the frequency shift was directly
ple: 650-1400 fSNPs to include animal strains. Through the correlated to the level of contamination (Pearson correla-
following sections, we used this tool to focus our analysis on tion coefficient=0.87). In contrast, for the remaining 1009
subtle, hard-to-detect changes introduced by contaminations. clean samples, no difference in frequency was observed (me-
dian=0%, IQR=0.6%), thus confirming again that removal
Contamination often leads to subtle errors in variant of non-MTBC reads does not impact the analysis in non-
calling that are elusive to conventional pipelines. Hav- contaminated samples.
ing established that contamination can have a large impact in
SNP detection, we compared the outcomes of WGS analysis Contamination can affect clinical predictions from
in the experimental dataset when using a conventional work- WGS data. To exemplify the extent of these alterations in
flow with the taxonomic filter approach described. Overall, WGS data interpretation, we evaluated how contamination
we found two main types of samples according to their level can impact drug resistance predictions and epidemiology.
of contamination. High proportions of contaminating reads When following the conventional analysis workflow, incor-
originate odd results such as very low sequencing depths, un- rect drug resistance susceptibility profiles (WGS-DST) were
even genome coverages, or extreme number of variants. This predicted for 53 samples according to known mutations de-
type of contamination is less likely to suppose a real threat, scribed in the PhyResSE catalog (Additional File 1). Among
as a careful analysis would probably detect it. Therefore, we these, 26 were false streptomycin resistances, 6 showed false
Goig et al. | Impact of contaminations in WGS bioRχiv | 3
bioRxiv preprint first posted online Aug. 29, 2018; doi: http://dx.doi.org/10.1101/403824. The copyright holder for this preprint
(which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity.
It is made available under a CC-BY-NC 4.0 International license.
resistance profiles to first line drugs and 1 sample showed an esized that removal of non-mycobacterial reads prior to map-
undetected resistance to both fluoroquinolones and strepto- ping might lead to the loss of several SNPs (23). Remark-
mycin. It must be noted that our pipeline reports any vSNP ably, the authors considered mycobacterial DNA as a whole,
in a frequency of at least the 10%. Since WGS-DST strongly disregarding non-tuberculosis mycobacteria. Here we show
depends on the frequency of the mutations and the cutoffs that both NTM and non-mycobacterial DNA can be found
used to call variable SNPs, we evaluated how contamination in cultures and primary diagnostic samples (Figure 1), which
introduced variant frequency fluctuations among resistance sequencing reads can be aligned along the MTBC reference
associated genes. Whereas no change was observed for clean genome (Figure 2a), leading to many false positive and neg-
samples (median=0%; IQR=0%), strong fluctuations were ative variant predictions (Figure 2b). We use both simulated
observed in the case of contaminated samples (median=18%; and real sequencing experiments to demonstrate that removal
IQR=43%). Strikingly, a large fraction of the vSNPs called of contaminant reads prior to mapping is a solid approach not
in these genes showed a frequency shift of at least the 20% affecting either the number of variants detected nor the fre-
as a consequence of contaminating reads (Figure 5a). quencies calculated. Of course, the efficacy of this method-
In order to assess the impact of contamination in epidemio- ology will directly depend on both the accuracy of the tax-
logical predictions, we used the fSNPs to calculate the pair- onomic classifier and the reference database used. In the
wise genetic distances for all samples belonging to the same case of MTB, Kraken showed an outstanding performance,
study. We observed that contamination strongly affected ge- although it was unable to distinguish some reads of M. avium,
netic distance calculations when comparing the conventional a closely related organism to the MTBC. In these cases, better
and the taxonomic filter pipelines (chi square < 0.01). Re- databases or slower alignment-based algorithms like BLAST
markably, the level of contamination did not strongly corre- could be used to improve the taxonomic filter accuracy.
late with the amount of change (Pearson correlation coeffi- While developing the pipeline to understand the impact of
cient=0.51), in agreement with the fact that slight contamina- contamination, we realized about the importance of the ref-
tions can be responsible for significant changes in the number erence genome chosen for mapping. Most bioinformatic
of SNPs, and vice versa. Contamination largely depends on pipelines for Mycobacterium tuberculosis align the sequenc-
sample source and laboratory procedures. This fact is clearly ing reads to the reference strain H37Rv. This strain has been
reflected in the amount of change for different types of sam- comprehensively studied since its isolation in 1905 and has
ple (Figure 5b). Genetic distance calculations are subject been historically used as reference genome as it was the first
to greater alterations in sputum sample sequencings than in Mycobacterium tuberculosis genome ever sequenced (24).
early MGIT culture sequencings (chi square < 0.01), and to H37Rv belongs to the lineage 4 of human MTBC and, as
greater alterations in the former than in standard cultures (chi a result, the variation expected when mapping to it deeply
square < 0.01). This distortion in genetic distance can lead to depends on how phylogenetically close is the strain under
define wrong epidemiological relationships. For instance, a study. In contrast, by mapping to an inferred ancestral MTBC
sample from Mozambique was unlinked from a transmission genome we are able to establish an expected range of SNPs
cluster due to a high number of contaminant reads. In this re- for any given MTB sample. We use this range as a tool for
gard, it is particularly interesting that some samples from the quality control, rapidly identifying extreme contaminations,
sputum capture-sequencing study showed several fSNPs of low quality sequencings, or super-infections with different
distance to their respective matching cultures, four of which strains.
were above typical transmission cutoffs (17, 19, 20 and 35
fSNPs respectively). When removing non-MTBC reads, all Using the taxonomic filter approach we estimate that around
genetic distances between sputum capture-sequencings and 94% of the variants identified in clinical samples from eight
their respective matching cultures were of 0 fSNPs. different WGS studies were introduced by contaminant reads.
Importantly, we show that many false SNPs can be called re-
gardless of the extent of contamination, with dozens of in-
Discussion correct calls even in samples that would normally be con-
In this work we use a taxonomic read classifier to check sidered as “pure”. All these alterations can have a massive
for contamination in an extensive dataset of M. tuberculo- impact in population diversity estimates, distorting the bio-
sis WGS samples, demonstrating that non-target reads can be logical conclusions that are drawn from WGS data. Studies
found in any type of sample, regardless of the experiment be- trying to estimate a molecular clock, or the action of selec-
ing conducted or the specimen source. The assumption that tive forces could be completely biased, since they are often
WGS from pure cultures only generates reads from the tar- based on the frequency spectrum derived from low frequency
get organism, and that non-target reads are barely mapped to variants (25). We exemplify this issue by comparing the out-
the reference genome, has encouraged pipelines vulnerable to comes of a conventional pipeline with a contamination-aware
contamination. Only few works included bioinformatic steps approach based on a taxonomic filter for two relevant appli-
aimed to deal with DNA contamination and, even in these cations: epidemiology and prediction of drug-resistant phe-
cases, the particular approaches used have not been evaluated notypes. When estimating transmission, we observed a con-
(13, 21, 22). siderable fSNPs-distance variation for many of the pairwise
A recent report focused on the artifactual variation in WGS distances calculated. This is of particular relevance in slow-
of Mycobacterium tuberculosis from MGIT cultures hypoth- evolving organisms like MTB, for which transmission events
4 | bioRχiv Goig et al. | Impact of contaminations in WGS
bioRxiv preprint first posted online Aug. 29, 2018; doi: http://dx.doi.org/10.1101/403824. The copyright holder for this preprint
(which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity.
It is made available under a CC-BY-NC 4.0 International license.
are determined based on few variants. Transmission estima- H37Rv (NC_000962.3) and the aforementioned MTBC an-
tion can be affected by either false positive or false negative cestral genome (20). We compared these two approaches
fSNPs, since contaminant reads can make the frequencies in terms of the number of fixed SNPs called for all non-
fluctuate around the threshold used to call fixed variants (90% contaminated samples, considering non-contaminated those
in this work). This holds true when performing WGS-DST, with more than the 99% of the reads classified as MTBC and
as predictions based on the observation of certain mutations producing at least 40X of median sequencing depth.
will finally depend on the thresholds used to call variable po-
sitions (10% in this work). Analysis pipeline: impact of contaminant DNA in vari-
Most of the contaminations and the errors they lead to are ant calling outcomes. In order to evaluate the impact of
very likely to pass unnoticed to conventional pipelines. We DNA contamination in WGS analysis, each sample was ana-
claim that inclusion of analysis steps aimed to deal with con- lyzed following three different methods. Firstly, each sam-
taminant data are indispensable in any bacterial WGS bioin- ple was analyzed using a conventional tuberculosis WGS
formatic workflow. This will be particularly relevant in large pipeline. In summary, reads were trimmed and filtered us-
sequencing projects directed towards the discovery of new ing Trimmomatic (31) to remove low-quality sequences and
genotype to phenotype associations and when performing di- then mapped to the MTBC ancestral genome using bwa mem
rect sequencing from complex clinical samples. (32). Variants were then called and filtered using VarScan2
(33) with two different set of parameters. To study transmis-
sion we used high-stringent parameters to obtain high con-
Methods fidence fixed variants (fSNPs) (minimum depth of 20 reads,
Experimental and simulated datasets analyzed. In or- average base quality of 20, p-value cutoff 0.01, observed in
der to evaluate the extent of contaminations among differ- both strands and minimum frequency of 90%), including re-
ent MTB WGS studies and their impact in the analysis out- moval of SNPs in repetitive and mobile regions, SNPs called
comes, we analyzed tuberculosis WGS runs from eight differ- within a 4bp window from deleted positions and high SNP
ent studies. Seven of these datasets were publicly available density regions (allowing a maximum of 3 SNPs in 10bp
beforehand (13, 14, 21, 26–29). Another dataset was gen- windows). In order to predict drug resistance and study bac-
erated in our laboratory, corresponding to an Illumina MiSeq terial subpopulations, we used another set of parameters that
sequencing of 138 samples from Mozambique see Additional allowed us to look into variable SNPs (vSNPs) (minimum
file 1. All these samples are referred as the experimental depth of 10 reads and variant observed in at least 6 reads, av-
dataset and comprised a total of 1553 Illumina runs (Table erage base quality of 20, p-value cutoff 0.01, observed in both
1). strands and minimum frequency of 10%), including removal
To validate our analysis and quantify the impact of contam- of SNPs in repetitive and mobile regions, and SNPs called
ination under controlled conditions, we generated a simu- near deleted positions within a window of 4bp. Sequencing
lated dataset. This dataset consisted of simulated Illumina depth was calculated for each sample using bedtools (34).
MiSeq 250bp paired-end sequencing experiments using ART Samples were then reanalyzed, following the same steps, but
(30) for the MTBC reference genome, the human genome adding a previous taxonomic filter removing those reads clas-
(GRCh38, Ensembl release 81) and 44 different non-MTBC sified by Kraken as any species other than Mycobacterium tu-
bacterial species (see Additional file 1). berculosis complex. Alternatively, samples were reanalyzed
substituting this taxonomic filter by a custom alignment fil-
ter. Different parameter combinations were tested to max-
Contamination assessment using Kraken. In order to imize removal of non-MTB mappings, minimizing loss of
assess the contamination level in each sample, all sequencing MTB mappings (see Additional file 1). The filter finally con-
reads were taxonomically classified using Kraken (15) with sisted in the removal of alignments with length, identity and
a custom database comprising all sequences of bacteria, ar- mapping quality below 40bp, 97% and 60 respectively.
chaea, virus, protozoa, plasmids and fungi in RefSeq (release
78), plus the human genome (GRCh38, Ensembl release 81). Estimation of false positive and negative calls in the
simulated dataset. In order to inspect which regions of the
Use of an ancestral genome to normalize the number reference genome are susceptible of recruiting non-MTBC
of SNPs. In this work we normalize the number of variants reads, we analyzed the samples of the simulated dataset as
for any given MTB sample by mapping to the inferred ances- described in the analysis pipeline and then measured the
tral genome of the MTBC, establishing a range of expected mean sequencing depth along the genome in 100 bp win-
SNPs that we use as a tool for quality control. To high- dows. To assess whether false positive SNPs and drug re-
light the hazard that unexpected contaminations suppose to sistance predictions are produced by these non-MTBC map-
WGS outcomes, we focus our analysis in samples present- pings, we generated mock contaminated samples by mixing
ing variations within this range that could, therefore, pass simulated sequencing reads from the MTBC ancestor refer-
completely unnoticed when using a conventional pipeline. ence with different proportions (5%, 15%, 30% and 70%) of
To better illustrate this, we analyzed all the samples from other organisms corresponding to 12 common contaminants
the experimental dataset as described in the variant calling identified in the experimental dataset. Therefore, any SNP
section mapping to both the conventional reference strain identified when analyzing these samples, would be a false
Goig et al. | Impact of contaminations in WGS bioRχiv | 5
bioRxiv preprint first posted online Aug. 29, 2018; doi: http://dx.doi.org/10.1101/403824. The copyright holder for this preprint
(which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity.
It is made available under a CC-BY-NC 4.0 International license.
positive SNP imputable to contamination. In addition, we 8. Christopher G. Wilson, Reuben W. Nowell, and Timothy G. Barraclough. Cross-
Contamination Explains "Inter and Intraspecific Horizontal Genetic Transfers" between
mapped these mock samples to a modified version of the ref- Asexual Bdelloid Rotifers. Current biology: CB, July 2018. ISSN 1879-0445. doi:
erence genome where we introduced two types of mutations. 10.1016/j.cub.2018.05.070.
9. Marion Ballenghien, Nicolas Faivre, and Nicolas Galtier. Patterns of cross-contamination in
Random mutations each 100bp, and all the drug resistance a multispecies population genomic project: detection, quantification, impact, and solutions.
conferring mutations described as “high confidence” in the BMC biology, 15(1):25, 2017. ISSN 1741-7007. doi: 10.1186/s12915-017-0366-6.
10. WORLD HEALTH ORGANIZATION. GLOBAL TUBERCULOSIS REPORT 2017. WORLD
PhyResSE catalog (19). Therefore, any of the introduced HEALTH ORGANIZATION, S.l., 2017. ISBN 978-92-4-156551-6. OCLC: 1017579392.
SNPs that were undetected when analyzing these samples, 11. Timothy M Walker, Camilla LC Ip, Ruth H Harrell, Jason T Evans, Georgia Kapatai, Martin J
Dedicoat, David W Eyre, Daniel J Wilson, Peter M Hawkey, Derrick W Crook, Julian Parkhill,
would be false negative SNPs attributable to contamination. David Harris, A Sarah Walker, Rory Bowden, Philip Monk, E Grace Smith, and Tim EA Peto.
Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retro-
spective observational study. The Lancet Infectious Diseases, 13(2):137–146, February
Pairwise genetic distances and drug resistance pre- 2013. ISSN 1473-3099. doi: 10.1016/S1473-3099(12)70277-3.
diction in the experimental dataset. For each study, ge- 12. Timothy M Walker, Thomas A Kohl, Shaheed V Omar, Jessica Hedge, Carlos Del Ojo Elias,
Phelim Bradley, Zamin Iqbal, Silke Feuerriegel, Katherine E Niehaus, Daniel J Wilson,
netic distances between samples were calculated from mul- David A Clifton, Georgia Kapatai, Camilla L C Ip, Rory Bowden, Francis A Drobniewski,
tiple alignments of non-redundant variable positions across Caroline Allix-Béguec, Cyril Gaudin, Julian Parkhill, Roland Diel, Philip Supply, Derrick W
Crook, E Grace Smith, A Sarah Walker, Nazir Ismail, Stefan Niemann, and Tim E A Peto.
samples using the R library APE (35) as the fSNP distance, Whole-genome sequencing for prediction of Mycobacterium tuberculosis drug susceptibil-
disregarding resistance-associated positions and using pair- ity and resistance: a retrospective cohort study. The Lancet Infectious Diseases, 15(10):
1193–1202, October 2015. ISSN 1473-3099. doi: 10.1016/S1473-3099(15)00062-6.
wise deletion for gap positions. Samples with at least 650 13. Amanda C. Brown, Josephine M. Bryant, Katja Einer-Jensen, Jolyon Holdstock, Darren T.
fixed SNPs to the MRCA genome and a median coverage of Houniet, Jacqueline Z. M. Chan, Daniel P. Depledge, Vladyslav Nikolayevskyy, Agnieszka
Broda, Madeline J. Stone, Mette T. Christiansen, Rachel Williams, Michael B. McAndrew,
20X were grouped in the same transmission cluster using a Helena Tutill, Julianne Brown, Mark Melzer, Caryn Rosmarin, Timothy D. McHugh, Robert J.
maximum distance threshold of 15 fSNPs to cover a range Shorten, Francis Drobniewski, Graham Speight, and Judith Breuer. Rapid Whole-Genome
Sequencing of Mycobacterium tuberculosis Isolates Directly from Clinical Samples. Journal
of recent and older transmission events. Drug resistance was of Clinical Microbiology, 53(7):2230–2237, July 2015. ISSN 0095-1137. doi: 10.1128/JCM.
predicted for samples with a median coverage of at least 20X 00486-15.
14. Antonina A. Votintseva, Phelim Bradley, Louise Pankhurst, Carlos del Ojo Elias, Matthew
according to the PhyResSE catalog of known resistance con- Loose, Kayzad Nilgiriwala, Anirvan Chatterjee, E. Grace Smith, Nicolas Sanderson, Tim-
ferring mutations. Mutations in resistance associated genes othy M. Walker, Marcus R. Morgan, David H. Wyllie, A. Sarah Walker, Tim E. A. Peto,
Derrick W. Crook, and Zamin Iqbal. Same-day diagnostic and surveillance data for tu-
that were not in this list and not described as phylogenetic berculosis via whole genome sequencing of direct respiratory samples. Journal of Clini-
polymorphisms in the literature, were annotated as “candi- cal Microbiology, pages JCM.02483–16, March 2017. ISSN 0095-1137, 1098-660X. doi:
10.1128/JCM.02483-16.
date resistance mutations” (see Additional file 1). 15. Derrick E. Wood and Steven L. Salzberg. Kraken: ultrafast metagenomic sequence classi-
fication using exact alignments. Genome Biology, 15:R46, March 2014. ISSN 1474-760X.
ACKNOWLEDGEMENTS doi: 10.1186/gb-2014-15-3-r46.
This work was funded by projects of the European Research Council (ERC) 16. Jørn A. Aas, Bruce J. Paster, Lauren N. Stokes, Ingar Olsen, and Floyd E. Dewhirst. Defining
(638553-TB-ACCELERATE) and Ministerio de Economia y Competitividad (Span- the Normal Bacterial Flora of the Oral Cavity. Journal of Clinical Microbiology, 43(11):5721–
ish Government) research grant SAF2016-77346-R (to IC), and BES-2014-071066 5732, November 2005. ISSN 0095-1137. doi: 10.1128/JCM.43.11.5721-5732.2005.
(to GAG). 17. Christine M. Bassis, John R. Erb-Downward, Robert P. Dickson, Christine M. Freeman,
Thomas M. Schmidt, Vincent B. Young, James M. Beck, Jeffrey L. Curtis, and Gary B.
AUTHOR CONTRIBUTIONS Huffnagle. Analysis of the Upper Respiratory Tract Microbiotas as the Source of the Lung
GAG and IC designed the study, analyzed the data and wrote the manuscript. AGB and Gastric Microbiotas in Healthy Individuals. mBio, 6(2), March 2015. ISSN 2150-7511.
and SB provided the samples from Mozambique. doi: 10.1128/mBio.00037-15.
COMPETING FINANCIAL INTERESTS 18. Stephen F. Altschul, Warren Gish, Webb Miller, Eugene W. Myers, and David J. Lipman.
Basic local alignment search tool. Journal of Molecular Biology, 215(3):403–410, October
The authors declare that they have no competing interests.
1990. ISSN 0022-2836. doi: 10.1016/S0022-2836(05)80360-2.
19. Silke Feuerriegel, Viola Schleusener, Patrick Beckert, Thomas A. Kohl, Paolo Miotto,
Daniela M. Cirillo, Andrea M. Cabibbe, Stefan Niemann, and Kurt Fellenberg. PhyResSE: a
Bibliography Web Tool Delineating Mycobacterium tuberculosis Antibiotic Resistance and Lineage from
Whole-Genome Sequencing Data. Journal of Clinical Microbiology, 53(6):1908–1914, June
1. Paul R McAdam, Emily J Richardson, and J Ross Fitzgerald. High-throughput sequencing 2015. ISSN 0095-1137. doi: 10.1128/JCM.00025-15.
for the study of bacterial pathogen biology. Current Opinion in Microbiology, 19:106–113, 20. Iñaki Comas, Jaidip Chakravartti, Peter M. Small, James Galagan, Stefan Niemann, Kristin
June 2014. ISSN 1369-5274. doi: 10.1016/j.mib.2014.06.002. Kremer, Joel D. Ernst, and Sebastien Gagneux. Human T cell epitopes of Mycobacterium
2. Xavier Didelot, Rory Bowden, Daniel J. Wilson, Tim E. A. Peto, and Derrick W. Crook. Trans- tuberculosis are evolutionarily hyperconserved. Nature Genetics, 42(6):498–503, June
forming clinical microbiology with bacterial genome sequencing. Nature reviews. Genetics, 2010. ISSN 1061-4036. doi: 10.1038/ng.590.
13(9):601–612, September 2012. ISSN 1471-0056. doi: 10.1038/nrg3226. 21. Louise J Pankhurst, Carlos del Ojo Elias, Antonina A Votintseva, Timothy M Walker, Kevin
3. David J. Roach, Joshua N. Burton, Choli Lee, Bethany Stackhouse, Susan M. Butler-Wu, Cole, Jim Davies, Jilles M Fermont, Deborah M Gascoyne-Binzi, Thomas A Kohl, Clare
Brad T. Cookson, Jay Shendure, and Stephen J. Salipante. A Year of Infection in the In- Kong, Nadine Lemaitre, Stefan Niemann, John Paul, Thomas R Rogers, Emma Roycroft,
tensive Care Unit: Prospective Whole Genome Sequencing of Bacterial Clinical Isolates E Grace Smith, Philip Supply, Patrick Tang, Mark H Wilcox, Sarah Wordsworth, David Wyllie,
Reveals Cryptic Transmissions and Novel Microbiota. PLOS Genetics, 11(7):e1005413, Li Xu, and Derrick W Crook. Rapid, comprehensive, and affordable mycobacterial diagnosis
July 2015. ISSN 1553-7404. doi: 10.1371/journal.pgen.1005413. with whole-genome sequencing: a prospective study. The Lancet. Respiratory Medicine, 4
4. Henk C. den Bakker, Marc W. Allard, Dianna Bopp, Eric W. Brown, John Fontana, Za- (1):49–58, January 2016. ISSN 2213-2600. doi: 10.1016/S2213-2600(15)00466-X.
min Iqbal, Aristea Kinney, Ronald Limberger, Kimberlee A. Musser, Matthew Shudt, Errol 22. José Afonso Guerra-Assunção, Rein M. G. J. Houben, Amelia C. Crampin, Themba
Strain, Martin Wiedmann, and William J. Wolfgang. Rapid Whole-Genome Sequencing for Mzembe, Kim Mallard, Francesc Coll, Palwasha Khan, Louis Banda, Arthur Chiwaya, Rui
Surveillance of Salmonella enterica Serovar Enteritidis. Emerging Infectious Diseases, 20 P. A. Pereira, Ruth McNerney, David Harris, Julian Parkhill, Taane G. Clark, and Judith R.
(8):1306–1314, August 2014. ISSN 1080-6040. doi: 10.3201/eid2008.131399. Glynn. Recurrence due to Relapse or Reinfection With Mycobacterium tuberculosis: A
5. Mette T. Christiansen, Amanda C. Brown, Samit Kundu, Helena J. Tutill, Rachel Williams, Whole-Genome Sequencing Approach in a Large, Population-Based Cohort With a High
Julianne R. Brown, Jolyon Holdstock, Martin J. Holland, Simon Stevenson, Jayshree HIV Infection Prevalence and Active Follow-up. The Journal of Infectious Diseases, 211(7):
Dave, CY William Tong, Katja Einer-Jensen, Daniel P. Depledge, and Judith Breuer. 1154–1163, April 2015. ISSN 0022-1899. doi: 10.1093/infdis/jiu574.
Whole-genome enrichment and sequencing of Chlamydia trachomatisdirectly from clini- 23. David H. Wyllie, Nicholas Sanderson, Richard Myers, Tim Peto, Esther Robinson, Derrick W.
cal samples. BMC Infectious Diseases, 14:591, November 2014. ISSN 1471-2334. doi: Crook, E. Grace Smith, and A. Sarah Walker. Control of artefactual variation in reported
10.1186/s12879-014-0591-3. inter-sample relatedness during clinical use of a Mycobacterium tuberculosis sequencing
6. Dhruba J. SenGupta, Lisa A. Cummings, Daniel R. Hoogestraat, Susan M. Butler-Wu, Jay pipeline. Journal of Clinical Microbiology, pages JCM.00104–18, June 2018. ISSN 0095-
Shendure, Brad T. Cookson, and Stephen J. Salipante. Whole-Genome Sequencing for 1137, 1098-660X. doi: 10.1128/JCM.00104-18.
High-Resolution Investigation of Methicillin-Resistant Staphylococcus aureus Epidemiology 24. S. T. Cole, R. Brosch, J. Parkhill, T. Garnier, C. Churcher, D. Harris, S. V. Gordon, K. Ei-
and Genome Plasticity. Journal of Clinical Microbiology, 52(8):2787–2796, January 2014. glmeier, S. Gas, C. E. Barry Iii, F. Tekaia, K. Badcock, D. Basham, D. Brown, T. Chill-
ISSN 0095-1137, 1098-660X. doi: 10.1128/JCM.00759-14. ingworth, R. Connor, R. Davies, K. Devlin, T. Feltwell, S. Gentles, N. Hamlin, S. Holroyd,
7. Richard W. Lusk. Diverse and widespread contamination evident in the unmapped depths T. Hornsby, K. Jagels, A. Krogh, J. McLean, S. Moule, L. Murphy, K. Oliver, J. Osborne,
of high throughput sequencing data. PloS One, 9(10):e110808, 2014. ISSN 1932-6203. M. A. Quail, M.-A. Rajandream, J. Rogers, S. Rutter, K. Seeger, J. Skelton, R. Squares,
doi: 10.1371/journal.pone.0110808. S. Squares, J. E. Sulston, K. Taylor, S. Whitehead, and B. G. Barrell. Deciphering the bi-
6 | bioRχiv Goig et al. | Impact of contaminations in WGS
bioRxiv preprint first posted online Aug. 29, 2018; doi: http://dx.doi.org/10.1101/403824. The copyright holder for this preprint
(which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity.
It is made available under a CC-BY-NC 4.0 International license.
ology of Mycobacterium tuberculosis from the complete genome sequence. Nature, 393
(6685):537–544, June 1998. ISSN 1476-4687. doi: 10.1038/31159.
25. Xavier Didelot, A. Sarah Walker, Tim E. Peto, Derrick W. Crook, and Daniel J. Wilson.
Within-host evolution of bacterial pathogens. Nature reviews. Microbiology, 14(3):150–162,
March 2016. ISSN 1740-1526. doi: 10.1038/nrmicro.2015.13.
26. Keira A. Cohen, Thomas Abeel, Abigail Manson McGuire, Christopher A. Desjardins, Van-
isha Munsamy, Terrance P. Shea, Bruce J. Walker, Nonkqubela Bantubani, Deepak V.
Almeida, Lucia Alvarado, Sinéad B. Chapman, Nomonde R. Mvelase, Eamon Y. Duffy,
Michael G. Fitzgerald, Pamla Govender, Sharvari Gujja, Susanna Hamilton, Clinton
Howarth, Jeffrey D. Larimer, Kashmeel Maharaj, Matthew D. Pearson, Margaret E. Priest,
Qiandong Zeng, Nesri Padayatchi, Jacques Grosset, Sarah K. Young, Jennifer Wortman,
Koleka P. Mlisana, Max R. O’Donnell, Bruce W. Birren, William R. Bishai, Alexander S.
Pym, and Ashlee M. Earl. Evolution of Extensively Drug-Resistant Tuberculosis over Four
Decades: Whole Genome Sequencing and Dating Analysis of Mycobacterium tuberculosis
Isolates from KwaZulu-Natal. PLOS Medicine, 12(9):e1001880, September 2015. ISSN
1549-1676. doi: 10.1371/journal.pmed.1001880.
27. Kurt R. Wollenberg, Christopher A. Desjardins, Aksana Zalutskaya, Vervara Slodovnikova,
Andrew J. Oler, Mariam Quiñones, Thomas Abeel, Sinead B. Chapman, Michael Tar-
takovsky, Andrei Gabrielian, Sven Hoffner, Aliaksandr Skrahin, Bruce W. Birren, Alexan-
der Rosenthal, Alena Skrahina, and Ashlee M. Earl. Whole-Genome Sequencing of My-
cobacterium tuberculosis Provides Insight into the Evolution and Genetic Composition of
Drug-Resistant Tuberculosis in Belarus. Journal of Clinical Microbiology, 55(2):457–469,
February 2017. ISSN 0095-1137. doi: 10.1128/JCM.02116-16.
28. Andrej Trauner, Qingyun Liu, Laura E. Via, Xin Liu, Xianglin Ruan, Lili Liang, Huimin Shi,
Ying Chen, Ziling Wang, Ruixia Liang, Wei Zhang, Wang Wei, Jingcai Gao, Gang Sun,
Daniela Brites, Kathleen England, Guolong Zhang, Sebastien Gagneux, Clifton E. Barry,
and Qian Gao. The within-host population dynamics of Mycobacterium tuberculosis vary
with treatment efficacy. Genome Biology, 18:71, April 2017. ISSN 1474-760X. doi: 10.
1186/s13059-017-1196-0.
29. Madikay Senghore, Jacob Otu, Adam Witney, Florian Gehre, Emma L. Doughty, Gemma L.
Kay, Phillip Butcher, Kayode Salako, Aderemi Kehinde, Nneka Onyejepu, Emmanuel Idigbe,
Tumani Corrah, Bouke de Jong, Mark J. Pallen, and Martin Antonio. Whole-genome se-
quencing illuminates the evolution and spread of multidrug-resistant tuberculosis in South-
west Nigeria. PLOS ONE, 12(9):e0184510, September 2017. ISSN 1932-6203. doi:
10.1371/journal.pone.0184510.
30. Weichun Huang, Leping Li, Jason R. Myers, and Gabor T. Marth. ART: a next-generation
sequencing read simulator. Bioinformatics, 28(4):593–594, February 2012. ISSN 1367-
4803. doi: 10.1093/bioinformatics/btr708.
31. Anthony M. Bolger, Marc Lohse, and Bjoern Usadel. Trimmomatic: a flexible trimmer for
Illumina sequence data. Bioinformatics, 30(15):2114–2120, August 2014. ISSN 1367-4803.
doi: 10.1093/bioinformatics/btu170.
32. Heng Li and Richard Durbin. Fast and accurate short read alignment with Burrows–Wheeler
transform. Bioinformatics, 25(14):1754–1760, July 2009. ISSN 1367-4803. doi: 10.1093/
bioinformatics/btp324.
33. Daniel C. Koboldt, Qunyuan Zhang, David E. Larson, Dong Shen, Michael D. McLellan,
Ling Lin, Christopher A. Miller, Elaine R. Mardis, Li Ding, and Richard K. Wilson. VarScan
2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing.
Genome Research, 22(3):568–576, March 2012. ISSN 1088-9051. doi: 10.1101/gr.129684.
111.
34. Aaron R. Quinlan. BEDTools: the Swiss-army tool for genome feature analysis. Current
protocols in bioinformatics / editoral board, Andreas D. Baxevanis ... [et al.], 47:11.12.1–
11.12.34, September 2014. ISSN 1934-3396. doi: 10.1002/0471250953.bi1112s47.
35. Emmanuel Paradis, Julien Claude, and Korbinian Strimmer. APE: Analyses of Phyloge-
netics and Evolution in R language. Bioinformatics, 20(2):289–290, January 2004. ISSN
1367-4803. doi: 10.1093/bioinformatics/btg412.
Goig et al. | Impact of contaminations in WGS bioRχiv | 7
bioRxiv preprint first posted online Aug. 29, 2018; doi: http://dx.doi.org/10.1101/403824. The copyright holder for this preprint
(which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity.
It is made available under a CC-BY-NC 4.0 International license.
Sputum Sputum
direct sequencing capture sequencing
Deep Nigeria Mozambique Direct Matching Capture Matching
sequencing Belarus KwaZulu MGIT sequencing MGIT sequencing 7H11
X an t ho mo na s
Ve illon ella
U n clas sif ie d
S t re pt o c o cc us
S t en ot rop ho mo na s
S t a p hy lo co c c us
Ro t hia
Rh od oc oc cu s
P s e ud op ro pion ib ac t eriu m
P se ud omon a s
Organism
P re vo t ella
P lu ra liba ct e r
P ae niba cillus
No n- T B- M y c o ba ct e ria
Ne ga t iv ic oc cu s
M TB C
M ic ro ba ct e rium
M et h an ot h ermo ba ct e r
M et h a n oc ulle us
La ct o co c c us
La ct o ba c illus
Kle b siella
Ho mo
G emella
E nt e ro c o cc us
Clos t ridioide s
C h iv ir us
Bif id ob ac t eriu m
Ba cillus
Ac t in omy c es
Ac in e t o ba ct e r
Percentage of sequencing reads
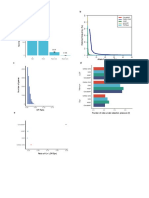
Fig. 1. Samples with contaminant sequencing reads are present in all the studies analyzed. Proportion of sequencing reads for different organisms across samples
from the experimental dataset. Each dot represents a sample with a given percentage of sequencing reads coming from the genus indicated in the y-axis. Only organisms in
a proportion above 2% are shown.
8 | bioRχiv Goig et al. | Impact of contaminations in WGS
bioRxiv preprint first posted online Aug. 29, 2018; doi: http://dx.doi.org/10.1101/403824. The copyright holder for this preprint
(which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity.
It is made available under a CC-BY-NC 4.0 International license.
Mycobacterial species Non mycobacterial species
Mean depth
a)
ndh katG pncA katG pncA
ndh
tlyA tlyA
rpsA rpsA
eis eis
0-5X
inhA inhA
fabG1 ahpC fabG1 ahpC
5-10X
16S 16S 10-15X
15-20X
thyA thyA
20-25X
25-30X
30-40X
40-50X
>50X
rplC rplC
rpsL rpsL
rpoB rpoB P. fluorescens
M. abscessus P. aeruginosa
M. smegmatis R. mucilaginosa
M. sinense B. dentium
M. fortuitum Propionibacterium sp.
M. phlei A. oris
ndhA ndhA
M. avium M. luteus
M. chimaera N. farcinica
M. haemophilum N. nova
M. kansasii N. brasiliensis
embC embC
embA embA
embB embB
gyrA ethA gyrA ethA
gyrB gidA gyrB gidA
b)
A. oris N. massiliensis P. fluorescens R. mucilaginosa S. aureus S. salivarius M. abscessus M. fortuitum M. kansasii M. chimaera M. avium
Conventional
10 5
10 4
Number of false SNPs
10 3
10 2
10
0
(log10 scale)
10 5
Mapping
10 4
filter
10 3
10 2
10
0
10 5
10 4 Taxonomic
filter
10 3
10 2
10
0
5%
%
%
%
5%
%
%
%
5%
%
%
%
5%
%
%
%
5%
%
%
%
5%
%
%
%
5%
%
%
%
5%
%
%
%
5%
%
%
%
5%
%
%
%
5%
%
%
%
15
30
70
15
30
70
15
30
70
15
30
70
15
30
70
15
30
70
15
30
70
15
30
70
15
30
70
15
30
70
15
30
70
False False Percentage of contaminant DNA
positives negatives
Fig. 2. Mapping of non-MTBC reads along the MTBC reference genome lead to false positive and negative calls. a) Mean sequencing depth obtained along the MTBC
reference genome across 100 bp windows when mapping 1,500,000 simulated reads of non-tuberculosis mycobacteria and organisms other than mycobacteria (OTM). The
top 10 organisms OTM that produced higher sequencing depths are shown. b) Number of false positive and negative SNPs (note log10 scale) called for mock samples
in-silico contaminated with different proportion of non-MTB organisms when following three different analysis pipelines (conventional, mapping filter and taxonomic filter). In
the case of the taxonomic filter, only contamination with M. avium produced false positive calls. Contamination with human DNA did not produce any false positive SNP
Goig et al. | Impact of contaminations in WGS bioRχiv | 9
bioRxiv preprint first posted online Aug. 29, 2018; doi: http://dx.doi.org/10.1101/403824. The copyright holder for this preprint
(which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity.
It is made available under a CC-BY-NC 4.0 International license.
Aligning to MTBC inferred ancestor Aligning to H37Rv reference strain
a) b) Lineage
Animal
Lineage 1
Lineage 2
Lineage 3
Number of fSNPs
Number of fSNPs
Lineage 4-4.6
Lineage 4.10
Lineage 5
Median sequencing depth Median sequencing depth
Fig. 3. Aligning to the inferred MTBC ancestor allows to define a range of expected SNPs for MTB samples. a) Number of fixed SNPs called for all samples of the
experimental dataset when using the MTBC inferred ancestor as reference genome. b) Number of fSNPs called when using the H37Rv strain as reference genome. Only
samples with more than 99% of MTBC reads are shown. Samples with median sequencing depths below 40X are shown only for illustrative purposes but were not considered
in the analysis.
Study
Belarus
Capture sequencing
Direct sequencing
KwaZulu
MGIT
Mozambique
Nigeria
Difference of SNPs
High depth
b
50% 60% 70% 80% 90% 100%
Percentage of MTBC
Fig. 4. Contamination is responsible for many false positive and negative SNPs in the experimental dataset. Difference between the number of SNPs called with the
conventional and the taxonomic filter pipelines. Each dot, representing a sample, is positioned in the x-axis according to the percentage of sequencing reads classified as
Mycobacterium tuberculosis Complex. a) False positive SNPs attributable to contamination. b) False negative SNPs attributable to contamination. Only samples with more
than 50% of MTBC and 650-1400 fSNPs to the MTBC MRCA reference are shown.
10 | bioRχiv Goig et al. | Impact of contaminations in WGS
bioRxiv preprint first posted online Aug. 29, 2018; doi: http://dx.doi.org/10.1101/403824. The copyright holder for this preprint
(which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity.
It is made available under a CC-BY-NC 4.0 International license.
a) b)
with a frequency fluctuation greater than 20%
100%
40%
Sputum sequencing
30%
Percentage of pairwise comparisons
90%
20%
Percentage of SNP calls
80%
10%
70% 0% p < 0.01
40%
60%
30%
Early MGIT
50%
20%
40% 10%
0%
30%
40% p < 0.01
Standard culture
20%
30%
10% 20%
10%
0%
0%
Pairwise SNP distance fluctuation
INH
INH/ETH
ETH
SM
SM/AMK/CPR/KAN
EMB
LZD
PAS
RMP
FQ
CPR
PZA
Fig. 5. Contaminant reads can lead to false clinical predictions. a) Fraction of vSNPs called within each drug-resistance associated gene that showed a frequency
shift greater than the 20% as a consequence of contamination. AMK (Amikacin); CPR (Capreomycin); EMB (Ethambutol); ETH (Ethionamide); FQ (Fluoroquinolones); INH
(Isoniazid); KAN (Kanamycin); LZD (Linezolid); PAS (Para-aminosalicylic acid); PZA (Pyrazinamide); RMP (Rifampicin); SM (Streptomycin). b)Distribution of fSNP distance
shifts introduced by contamination for each type of sample. Negative fluctuations correspond to false negative fSNPs of distance as a consequence of contamination, whereas
positive fluctuations correspond to false positive fSNPs
Goig et al. | Impact of contaminations in WGS bioRχiv | 11
bioRxiv preprint first posted online Aug. 29, 2018; doi: http://dx.doi.org/10.1101/403824. The copyright holder for this preprint
(which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity.
It is made available under a CC-BY-NC 4.0 International license.
Table 1. Studies of the experimental dataset. *This study included sequencings from non-MTB organisms. We analyzed the 168 reported as MTB by the authors.
Study Publication Runs analyzed Accession
Mozambique Unpublished 138 PRJEB27421
Kwazulu-Natal Cohen et al. 2015 433 PRJNA183624
Nigeria Senghore et al. 2017 73 PRJEB15857
Belarus Wollenberg et al. 2017 552 PRJNA200335
High depth Trauner et al. 2017 63 PRJEB13325, PRJEB17864
Sputum capture-sequencing Brown et al. 2015 58 PRJEB9206
Sputum direct-sequencing Votintseva et al. 2017 68 SRP093599
MGIT sequencing Pankhurst et al. 2016 168* PRJNA268101, PRJNA302362
12 | bioRχiv Goig et al. | Impact of contaminations in WGS
Das könnte Ihnen auch gefallen
- The Yellow House: A Memoir (2019 National Book Award Winner)Von EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Bewertung: 4 von 5 Sternen4/5 (98)
- Supplementary Table S13. Analysis of Important Snps Related To Alcohol and Caffeine MetabolismDokument6 SeitenSupplementary Table S13. Analysis of Important Snps Related To Alcohol and Caffeine MetabolismkjhgfghjkNoch keine Bewertungen
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeVon EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeBewertung: 4 von 5 Sternen4/5 (5795)
- Supplementary Table S9. Distribution of Pathagenic Variants Associated With HypothyroidismDokument2 SeitenSupplementary Table S9. Distribution of Pathagenic Variants Associated With HypothyroidismkjhgfghjkNoch keine Bewertungen
- Shoe Dog: A Memoir by the Creator of NikeVon EverandShoe Dog: A Memoir by the Creator of NikeBewertung: 4.5 von 5 Sternen4.5/5 (537)
- Supplementary Table S8. Disease-Causing Variants in Pancreatitis and Hypothyroidism From The ChinamapDokument21 SeitenSupplementary Table S8. Disease-Causing Variants in Pancreatitis and Hypothyroidism From The ChinamapkjhgfghjkNoch keine Bewertungen
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureVon EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureBewertung: 4.5 von 5 Sternen4.5/5 (474)
- Supplementary Table S11. The Significant Single Variants Associated With BMIDokument3 SeitenSupplementary Table S11. The Significant Single Variants Associated With BMIkjhgfghjkNoch keine Bewertungen
- Grit: The Power of Passion and PerseveranceVon EverandGrit: The Power of Passion and PerseveranceBewertung: 4 von 5 Sternen4/5 (588)
- Supplementary Table S6. Number of Autosome Loss-Of-Function Variants in The ChinamapDokument4 SeitenSupplementary Table S6. Number of Autosome Loss-Of-Function Variants in The ChinamapkjhgfghjkNoch keine Bewertungen
- On Fire: The (Burning) Case for a Green New DealVon EverandOn Fire: The (Burning) Case for a Green New DealBewertung: 4 von 5 Sternen4/5 (74)
- Supplementary Table S5. Number of Protein-Coding Genes Affected by Loss-Of-Function Variants With An Allele Frequency of 1%Dokument2 SeitenSupplementary Table S5. Number of Protein-Coding Genes Affected by Loss-Of-Function Variants With An Allele Frequency of 1%kjhgfghjkNoch keine Bewertungen
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryVon EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryBewertung: 3.5 von 5 Sternen3.5/5 (231)
- Supplementary Table S4. Number of Novel Autosome Variants Grouped by Impact and FrequencyDokument2 SeitenSupplementary Table S4. Number of Novel Autosome Variants Grouped by Impact and FrequencykjhgfghjkNoch keine Bewertungen
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceVon EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceBewertung: 4 von 5 Sternen4/5 (895)
- 2020 322 Moesm7 EsmDokument2 Seiten2020 322 Moesm7 EsmkjhgfghjkNoch keine Bewertungen
- Never Split the Difference: Negotiating As If Your Life Depended On ItVon EverandNever Split the Difference: Negotiating As If Your Life Depended On ItBewertung: 4.5 von 5 Sternen4.5/5 (838)
- Supplementary Table S1. Geographical Distribution of The Participants in The ChinamapDokument2 SeitenSupplementary Table S1. Geographical Distribution of The Participants in The ChinamapkjhgfghjkNoch keine Bewertungen
- The Little Book of Hygge: Danish Secrets to Happy LivingVon EverandThe Little Book of Hygge: Danish Secrets to Happy LivingBewertung: 3.5 von 5 Sternen3.5/5 (400)
- Supplementary Table S2. The Distribution and Number of Autosome Variants Grouped by Impact and FrequencyDokument3 SeitenSupplementary Table S2. The Distribution and Number of Autosome Variants Grouped by Impact and FrequencykjhgfghjkNoch keine Bewertungen
- Regulatory T Cells and Human Disease: Annual Review of ImmunologyDokument29 SeitenRegulatory T Cells and Human Disease: Annual Review of ImmunologykjhgfghjkNoch keine Bewertungen
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersVon EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersBewertung: 4.5 von 5 Sternen4.5/5 (345)
- Supplementary Table S3. Summary Statics of Variants Identified in The ChinamapDokument2 SeitenSupplementary Table S3. Summary Statics of Variants Identified in The ChinamapkjhgfghjkNoch keine Bewertungen
- 2020 322 Moesm6 EsmDokument2 Seiten2020 322 Moesm6 EsmkjhgfghjkNoch keine Bewertungen
- The Unwinding: An Inner History of the New AmericaVon EverandThe Unwinding: An Inner History of the New AmericaBewertung: 4 von 5 Sternen4/5 (45)
- Number of Indels (×100K) Frequency of ChinamapDokument2 SeitenNumber of Indels (×100K) Frequency of ChinamapkjhgfghjkNoch keine Bewertungen
- Team of Rivals: The Political Genius of Abraham LincolnVon EverandTeam of Rivals: The Political Genius of Abraham LincolnBewertung: 4.5 von 5 Sternen4.5/5 (234)
- 2020 322 Moesm4 EsmDokument2 Seiten2020 322 Moesm4 EsmkjhgfghjkNoch keine Bewertungen
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyVon EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyBewertung: 3.5 von 5 Sternen3.5/5 (2259)
- Bioinformatic Prediction of Potential T Cell Epitopes For SARS-Cov-2Dokument7 SeitenBioinformatic Prediction of Potential T Cell Epitopes For SARS-Cov-2kjhgfghjkNoch keine Bewertungen
- chr1 chr2 chr3 chr4: Position (MB)Dokument2 Seitenchr1 chr2 chr3 chr4: Position (MB)kjhgfghjkNoch keine Bewertungen
- C HR Omosome Structure at Micro-Scale: Functional Interplay Between Transcription and Fine-Scale Chromatin StructureDokument1 SeiteC HR Omosome Structure at Micro-Scale: Functional Interplay Between Transcription and Fine-Scale Chromatin StructurekjhgfghjkNoch keine Bewertungen
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaVon EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaBewertung: 4.5 von 5 Sternen4.5/5 (266)
- Observation Method in Qualitative ResearchDokument42 SeitenObservation Method in Qualitative Researchsantoshipoudel08Noch keine Bewertungen
- The Emperor of All Maladies: A Biography of CancerVon EverandThe Emperor of All Maladies: A Biography of CancerBewertung: 4.5 von 5 Sternen4.5/5 (271)
- Tips Experiments With MatlabDokument190 SeitenTips Experiments With MatlabVishalNoch keine Bewertungen
- Prelim Examination in Logic DesignDokument5 SeitenPrelim Examination in Logic Designdj_eqc7227Noch keine Bewertungen
- MAK Halliday The Language of ScienceDokument268 SeitenMAK Halliday The Language of ScienceJeneiGabriellaOfő100% (4)
- 1st Grading Exam Perdev With TosDokument4 Seiten1st Grading Exam Perdev With Toshazel malaga67% (3)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreVon EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreBewertung: 4 von 5 Sternen4/5 (1090)
- Curriculum Vitae: Name: Father S Name: Cell No: Email: Date of Birth: Nationality: Cnic No: Passport No: AddressDokument2 SeitenCurriculum Vitae: Name: Father S Name: Cell No: Email: Date of Birth: Nationality: Cnic No: Passport No: AddressJahanzeb JojiNoch keine Bewertungen
- (Solved) at The Instant Shown, 60Dokument3 Seiten(Solved) at The Instant Shown, 60marcosbispolimaNoch keine Bewertungen
- 2pages 6Dokument3 Seiten2pages 6Rachana G Y Rachana G YNoch keine Bewertungen
- Niagara AX Browser Access GuideDokument84 SeitenNiagara AX Browser Access GuideccitarellaNoch keine Bewertungen
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Von EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Bewertung: 4.5 von 5 Sternen4.5/5 (121)
- IBRO News 2004Dokument8 SeitenIBRO News 2004International Brain Research Organization100% (1)
- Solarbotics Wheel Watcher Encoder ManualDokument10 SeitenSolarbotics Wheel Watcher Encoder ManualYash SharmaNoch keine Bewertungen
- Mathematics ExcerciseDokument74 SeitenMathematics Excerciseserge129100% (1)
- Healthcare ProfessionalismDokument28 SeitenHealthcare ProfessionalismAnjo CincoNoch keine Bewertungen
- Chris Barnes UEFA Presentation Nov 2015Dokument47 SeitenChris Barnes UEFA Presentation Nov 2015muller.patrik1Noch keine Bewertungen
- My Home, My School: Meet and GreetDokument2 SeitenMy Home, My School: Meet and GreetAcebuque Arby Immanuel GopeteoNoch keine Bewertungen
- Practice Makes Perfect English Conversation UNIT 9Dokument10 SeitenPractice Makes Perfect English Conversation UNIT 9Thaís C C MirandaNoch keine Bewertungen
- Policy Analysis ReportDokument16 SeitenPolicy Analysis ReportGhelvin Auriele AguirreNoch keine Bewertungen
- Fendrick Govt 720 Global Problem EssayDokument5 SeitenFendrick Govt 720 Global Problem Essayapi-283893191Noch keine Bewertungen
- MRP Configuration GuideDokument12 SeitenMRP Configuration Guideofaofa1Noch keine Bewertungen
- Her Body and Other Parties: StoriesVon EverandHer Body and Other Parties: StoriesBewertung: 4 von 5 Sternen4/5 (821)
- Math Internal Assessment:: Investigating Parametric Equations in Motion ProblemsDokument16 SeitenMath Internal Assessment:: Investigating Parametric Equations in Motion ProblemsByron JimenezNoch keine Bewertungen
- Joshi C M, Vyas Y - Extensions of Certain Classical Integrals of Erdélyi For Gauss Hypergeometric Functions - J. Comput. and Appl. Mat. 160 (2003) 125-138Dokument14 SeitenJoshi C M, Vyas Y - Extensions of Certain Classical Integrals of Erdélyi For Gauss Hypergeometric Functions - J. Comput. and Appl. Mat. 160 (2003) 125-138Renee BravoNoch keine Bewertungen
- Vertical Axis Wind Turbine ProjDokument2 SeitenVertical Axis Wind Turbine Projmacsan sanchezNoch keine Bewertungen
- TASK 500 Technical Memorandum No. 503 Review and Assessment of Drainage Control Policies, Procedures, AND Guidelines Final DraftDokument31 SeitenTASK 500 Technical Memorandum No. 503 Review and Assessment of Drainage Control Policies, Procedures, AND Guidelines Final DraftGreg BraswellNoch keine Bewertungen
- B.tech - Non Credit Courses For 2nd Year StudentsDokument4 SeitenB.tech - Non Credit Courses For 2nd Year StudentsNishant MishraNoch keine Bewertungen
- SN Conceptual & Strategic SellingDokument34 SeitenSN Conceptual & Strategic Sellingayushdixit100% (1)
- CBSE Class 10 Science Sample Paper-08 (For 2013)Dokument11 SeitenCBSE Class 10 Science Sample Paper-08 (For 2013)cbsesamplepaperNoch keine Bewertungen
- 2.2 Lesson Notes FA12Dokument24 Seiten2.2 Lesson Notes FA12JAPNoch keine Bewertungen
- A Theory of Ethnic Group BoundariesDokument28 SeitenA Theory of Ethnic Group BoundariesAbdellatif El BekkariNoch keine Bewertungen
- A Tribute To Fernando L. L. B. Carneiro (1913 - 2001)Dokument2 SeitenA Tribute To Fernando L. L. B. Carneiro (1913 - 2001)Roberto Urrutia100% (1)
- Wifi HackDokument20 SeitenWifi HackVAJIR ABDURNoch keine Bewertungen
- Do You Believe in Magic?: The Sense and Nonsense of Alternative MedicineVon EverandDo You Believe in Magic?: The Sense and Nonsense of Alternative MedicineNoch keine Bewertungen
- CISM Certified Information Security Manager Study GuideVon EverandCISM Certified Information Security Manager Study GuideNoch keine Bewertungen
- Uncontrolled Spread: Why COVID-19 Crushed Us and How We Can Defeat the Next PandemicVon EverandUncontrolled Spread: Why COVID-19 Crushed Us and How We Can Defeat the Next PandemicNoch keine Bewertungen