Das könnte Ihnen auch gefallen
- The Spectra and Dynamics of Diatomic Molecules: Revised and Enlarged EditionVon EverandThe Spectra and Dynamics of Diatomic Molecules: Revised and Enlarged EditionNoch keine Bewertungen
- Equalizer Design ExampleDokument10 SeitenEqualizer Design ExamplehaimanhptitNoch keine Bewertungen
- EjercicosDokument8 SeitenEjercicosdavidNoch keine Bewertungen
- Distributed Model Predictive Control for Plant-Wide SystemsVon EverandDistributed Model Predictive Control for Plant-Wide SystemsNoch keine Bewertungen
- 130nm BulkDokument3 Seiten130nm Bulkunseen worldNoch keine Bewertungen
- Modern Borehole Analytics: Annular Flow, Hole Cleaning, and Pressure ControlVon EverandModern Borehole Analytics: Annular Flow, Hole Cleaning, and Pressure ControlNoch keine Bewertungen
- Ece 346 Lab 4Dokument6 SeitenEce 346 Lab 4LordLawnmowerNoch keine Bewertungen
- Tabla de Datos Experimentales Experimento Tiempo (Min) Vol. HCL (ML) 1 2 3 4 5 6 7 8 9 10Dokument4 SeitenTabla de Datos Experimentales Experimento Tiempo (Min) Vol. HCL (ML) 1 2 3 4 5 6 7 8 9 10DenisRogerNoch keine Bewertungen
- DAD1 A, Sig 254,4 Ref Off (CHEM112 - 211 - 7S - A7.D)Dokument100 SeitenDAD1 A, Sig 254,4 Ref Off (CHEM112 - 211 - 7S - A7.D)Ucb BioeNoch keine Bewertungen
- CH 3 Part 2 Tutorial - May20Dokument15 SeitenCH 3 Part 2 Tutorial - May20Scorpion RoyalNoch keine Bewertungen
- HW2 Ans RevDokument14 SeitenHW2 Ans RevanonymoussionNoch keine Bewertungen
- Superintend by D.foad: Ministry of Higher Education and Scientific ResearchDokument7 SeitenSuperintend by D.foad: Ministry of Higher Education and Scientific Researchali najatNoch keine Bewertungen
- Fender Base Plate Analysis: Static CaseDokument7 SeitenFender Base Plate Analysis: Static CaseBhavesh ShiyaniNoch keine Bewertungen
- Equation Solving ExamplesDokument10 SeitenEquation Solving Examplesalif08042012Noch keine Bewertungen
- Equation Solving ExamplesDokument10 SeitenEquation Solving Examplesalif08042012Noch keine Bewertungen
- GMM FinalDokument7 SeitenGMM FinalIntan Masyitha IrwanNoch keine Bewertungen
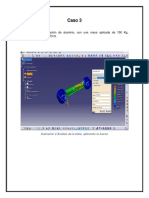
- Caso 3Dokument9 SeitenCaso 3ramiroNoch keine Bewertungen
- An Example of The Backward Elimination ProcessDokument8 SeitenAn Example of The Backward Elimination ProcessMohammedseid AhmedinNoch keine Bewertungen
- Econometrics Hina NazDokument17 SeitenEconometrics Hina NazHina Naz (F-Name :Khadim Hussain)Noch keine Bewertungen
- Zub Indeterminate Truss DegDokument13 SeitenZub Indeterminate Truss DegAnonymous jmo0bcjAsNoch keine Bewertungen
- Solutions Manual To Accompany A Second Course in Statistics Regression Analysis 7th Edition 0321691695Dokument47 SeitenSolutions Manual To Accompany A Second Course in Statistics Regression Analysis 7th Edition 0321691695RhondaCookqjdyg100% (84)
- EE115 Solution To Prob 3 and 5 Per Unit AnalysisDokument12 SeitenEE115 Solution To Prob 3 and 5 Per Unit AnalysisJoshua Roberto GrutaNoch keine Bewertungen
- Negara Maju UpdDokument10 SeitenNegara Maju UpdKapibaraNoch keine Bewertungen
- MuhammadZahranFadhillah - 1167030050 - Tugas Mandiri KomtelDokument8 SeitenMuhammadZahranFadhillah - 1167030050 - Tugas Mandiri KomtelMohammad Zaky AzhariNoch keine Bewertungen
- 180 NM PTM BSIM3Dokument3 Seiten180 NM PTM BSIM3Kshitij Goel0% (1)
- Lab 1Dokument7 SeitenLab 1Kashif hussainNoch keine Bewertungen
- Long Report Exp 6Dokument6 SeitenLong Report Exp 6Mxokzah Cmoh100% (1)
- FeareportDokument7 SeitenFeareportapi-404637171Noch keine Bewertungen
- Linear Programming - Simplex Vs Interior-2-10Dokument9 SeitenLinear Programming - Simplex Vs Interior-2-10Fan ClubNoch keine Bewertungen
- AS Practice Exercise 3 Model Solution PDFDokument2 SeitenAS Practice Exercise 3 Model Solution PDFRohan KanungoNoch keine Bewertungen
- PB Mod 2 1 Homework 1 LL 2016 HRDokument8 SeitenPB Mod 2 1 Homework 1 LL 2016 HRAntonio ChissanoNoch keine Bewertungen
- Aa + BB +CC+DD RR A+ (B/a) B + (C/a) C + (D/a) D (R/a) R R - KC C C C N N (1-X) N N - (B/a) (N - N) N N - (C/a) (N - N) N N - (D/a) (N - N) C N /VDokument7 SeitenAa + BB +CC+DD RR A+ (B/a) B + (C/a) C + (D/a) D (R/a) R R - KC C C C N N (1-X) N N - (B/a) (N - N) N N - (C/a) (N - N) N N - (D/a) (N - N) C N /VChemical EngineeringNoch keine Bewertungen
- Tugas PotensiometriDokument4 SeitenTugas PotensiometriNandia Salsa Restu MulyaniNoch keine Bewertungen
- Ministry of Higher Education and Scientific Research: Kirkuk Civil Engineering Stage - ThreeDokument8 SeitenMinistry of Higher Education and Scientific Research: Kirkuk Civil Engineering Stage - Threeali najatNoch keine Bewertungen
- Metode Linear BergandaDokument16 SeitenMetode Linear BergandaEka Fatihatul HikmahNoch keine Bewertungen
- OTK KristiantyDokument9 SeitenOTK KristiantyDewi RatnasariNoch keine Bewertungen
- Nmos180 LibDokument2 SeitenNmos180 LibKaramveer SinghNoch keine Bewertungen
- EXAMPLE 8.32: N-HeptaneDokument7 SeitenEXAMPLE 8.32: N-HeptaneAria DarmawanNoch keine Bewertungen
- Id Sepallengthcm Sepalwidthcm Petallengthcm Petalwidthcm Species 0 1 2 3 4Dokument4 SeitenId Sepallengthcm Sepalwidthcm Petallengthcm Petalwidthcm Species 0 1 2 3 4Sebastian CallesNoch keine Bewertungen
- Ejercicio Numero 4Dokument8 SeitenEjercicio Numero 4natanael quiñoneNoch keine Bewertungen
- Assignment 2.solutionDokument13 SeitenAssignment 2.solutionteweldeNoch keine Bewertungen
- Machine Design ProblemsDokument10 SeitenMachine Design ProblemsSiddharth ShekharNoch keine Bewertungen
- Rotación de PavimentoDokument7 SeitenRotación de PavimentoIván Pimentel OrtegaNoch keine Bewertungen
- Caso 1Dokument9 SeitenCaso 1ramiroNoch keine Bewertungen
- Impedance and Reactance: Faculty of Science Department of Physics Physics Lab 112Dokument4 SeitenImpedance and Reactance: Faculty of Science Department of Physics Physics Lab 112issa alatrashNoch keine Bewertungen
- TekresDokument3 SeitenTekresMohammad Rayhan Kim Abdul SalamNoch keine Bewertungen
- T2 3 PDFDokument3 SeitenT2 3 PDFTravis BickleNoch keine Bewertungen
- UntitledDokument8 SeitenUntitledLucas Hernández Karla BereniceNoch keine Bewertungen
- Component y 1. Apparent Molecular Weight (Ma) Mi Yi C1 C2 C3 n-C4 n-C5 C6 P C7+ T Jumlah RDokument26 SeitenComponent y 1. Apparent Molecular Weight (Ma) Mi Yi C1 C2 C3 n-C4 n-C5 C6 P C7+ T Jumlah RClaviano LeiwakabessyNoch keine Bewertungen
- QTBDokument7 SeitenQTBUmarNoch keine Bewertungen
- Applied Numerical Methods Wmatlab For Engineers and Scientists 3rd Edition Chapra Solutions ManualDokument25 SeitenApplied Numerical Methods Wmatlab For Engineers and Scientists 3rd Edition Chapra Solutions ManualJosephCraiggmax100% (52)
- Dwnload Full Applied Numerical Methods Wmatlab For Engineers and Scientists 3rd Edition Chapra Solutions Manual PDFDokument36 SeitenDwnload Full Applied Numerical Methods Wmatlab For Engineers and Scientists 3rd Edition Chapra Solutions Manual PDFeyepieceinexact.4h0h100% (11)
- Tristan Camilleri - SOR0511 - Questions - 2021.06.20Dokument33 SeitenTristan Camilleri - SOR0511 - Questions - 2021.06.20TristanCamilleriNoch keine Bewertungen
- יניבDokument9 SeitenיניבramikNoch keine Bewertungen
- Natural Gas Homework2Dokument42 SeitenNatural Gas Homework2Khanz KhanNoch keine Bewertungen
- Final Group Report Exp 2 A4Dokument5 SeitenFinal Group Report Exp 2 A4nonononowayNoch keine Bewertungen
- Structural Design of Two Storey Recidencnce Owner: Ferdinand Dela Cruz Address: Sto. Tomas, San Jose CityDokument11 SeitenStructural Design of Two Storey Recidencnce Owner: Ferdinand Dela Cruz Address: Sto. Tomas, San Jose CityVic ValdezNoch keine Bewertungen
- Superintend by D.foad: Ministry of Higher Education and Scientific ResearchDokument7 SeitenSuperintend by D.foad: Ministry of Higher Education and Scientific ResearchAhmed falahNoch keine Bewertungen
- Sio 2Dokument6 SeitenSio 2Missiles dodgedNoch keine Bewertungen
- Materials Letters: Ovidiu Chiscan, Ioan Dumitru, Petronel Postolache, Vasile Tura, Alexandru StancuDokument4 SeitenMaterials Letters: Ovidiu Chiscan, Ioan Dumitru, Petronel Postolache, Vasile Tura, Alexandru StancuMissiles dodgedNoch keine Bewertungen
- Microwave Absorptions of Ultrathin Conductive Films and Designs of Frequency-Independent Ultrathin AbsorbersDokument7 SeitenMicrowave Absorptions of Ultrathin Conductive Films and Designs of Frequency-Independent Ultrathin AbsorbersMissiles dodgedNoch keine Bewertungen
- Greenhouse Gas Emissions From A Typical Passenger VehicleDokument5 SeitenGreenhouse Gas Emissions From A Typical Passenger VehicleMissiles dodgedNoch keine Bewertungen
- Nuclear Research ArticleDokument8 SeitenNuclear Research ArticleMissiles dodgedNoch keine Bewertungen
- Rails BasicsDokument229 SeitenRails BasicsachhuNoch keine Bewertungen
- Modern Age Waste Water ProblemsDokument364 SeitenModern Age Waste Water Problemsromaehab201912Noch keine Bewertungen
- Valve & Amplifier Design, Valve EquivalentsDokument51 SeitenValve & Amplifier Design, Valve EquivalentsValve Data80% (5)
- 4july BookDokument5 Seiten4july BookDevansh AggarwalNoch keine Bewertungen
- System Administration JakartaDokument347 SeitenSystem Administration JakartaLorena Castillero80% (10)
- PDF Sesion de Aprendizaje de Comunicacion Leemos y Cantamos Canciones Criollas Lambayecanas - CompressDokument6 SeitenPDF Sesion de Aprendizaje de Comunicacion Leemos y Cantamos Canciones Criollas Lambayecanas - CompressJulia Navarro CheroNoch keine Bewertungen
- Microstructure Characteristics and Performance of Dissimilar Welds Between Magnesium Alloy and Aluminum Formed by Friction StirringDokument5 SeitenMicrostructure Characteristics and Performance of Dissimilar Welds Between Magnesium Alloy and Aluminum Formed by Friction StirringLeidy Silvana Chacón VelascoNoch keine Bewertungen
- 07 Bubble BreakDokument25 Seiten07 Bubble BreakWeb LogueandoNoch keine Bewertungen
- Forces Revision Questions 1. Resistive Force.: Island School 1Dokument13 SeitenForces Revision Questions 1. Resistive Force.: Island School 1Deepal PrasankaNoch keine Bewertungen
- 2018 06 OnlineDokument12 Seiten2018 06 OnlineMohamed HasikNoch keine Bewertungen
- Analytic Geometry Parabola ProblemsDokument14 SeitenAnalytic Geometry Parabola ProblemsOjit QuizonNoch keine Bewertungen
- GGGB6023 Tugasan Tutorial 3 - P69060 Mior SyazrilDokument5 SeitenGGGB6023 Tugasan Tutorial 3 - P69060 Mior SyazrilAmizan AbdullahNoch keine Bewertungen
- Caliper Xy MemoryDokument6 SeitenCaliper Xy MemoryA MuNoch keine Bewertungen
- Ubd Planning Template With QuestionsDokument3 SeitenUbd Planning Template With Questionsapi-217297849Noch keine Bewertungen
- EWAD-CF EEDEN15-435 Data Books EnglishDokument42 SeitenEWAD-CF EEDEN15-435 Data Books EnglishrpufitaNoch keine Bewertungen
- TH 2100Dokument67 SeitenTH 2100KI TechnologiesNoch keine Bewertungen
- UNIT1-Demand Management in Supply Chain Demand Planning and ForecastingDokument20 SeitenUNIT1-Demand Management in Supply Chain Demand Planning and Forecastingshenbha50% (2)
- Power Off Reset Reason BackupDokument5 SeitenPower Off Reset Reason Backupmohamed ahmedNoch keine Bewertungen
- Numerical Methods in Rock Mechanics - 2002 - International Journal of Rock Mechanics and Mining SciencesDokument19 SeitenNumerical Methods in Rock Mechanics - 2002 - International Journal of Rock Mechanics and Mining SciencesAnderson Lincol Condori PaytanNoch keine Bewertungen
- Fire InvestigationDokument126 SeitenFire InvestigationAbcede IloiloNoch keine Bewertungen
- API-650 Design Procedure ExampleDokument21 SeitenAPI-650 Design Procedure Examplegdwvcd93% (14)
- AMC2019 StudentsResults Indonesia 8JDokument4 SeitenAMC2019 StudentsResults Indonesia 8JWinety Kristiana DewiNoch keine Bewertungen
- Vector AlgebraDokument7 SeitenVector AlgebraDeeeNoch keine Bewertungen
- HP Proliant DL380 G6 Server - Step by StepDokument9 SeitenHP Proliant DL380 G6 Server - Step by StepBoss100% (1)
- Condensation and BoilingDokument14 SeitenCondensation and BoilingCrislyn Akilit Bayawa100% (1)
- Calculation Sheet Boiler Control BuildingDokument35 SeitenCalculation Sheet Boiler Control BuildingKhamal Rachmanda AdamNoch keine Bewertungen
- 3DC Real Light 24HDRi Vol03Dokument27 Seiten3DC Real Light 24HDRi Vol03AntezanaFernandoNoch keine Bewertungen
- Adjectives 4Dokument34 SeitenAdjectives 4Delia Bolasoc100% (1)
- Tugas HKSA Deskriptor (Fitriani Choerunnisa (11171013) 3FA1)Dokument4 SeitenTugas HKSA Deskriptor (Fitriani Choerunnisa (11171013) 3FA1)fitriani choerunnisaNoch keine Bewertungen
- Unit Iv Ce 6405Dokument13 SeitenUnit Iv Ce 6405HanafiahHamzahNoch keine Bewertungen
- Summary and Interpretation of Reality TransurfingVon EverandSummary and Interpretation of Reality TransurfingBewertung: 5 von 5 Sternen5/5 (5)
- A Brief History of Time: From the Big Bang to Black HolesVon EverandA Brief History of Time: From the Big Bang to Black HolesBewertung: 4 von 5 Sternen4/5 (2193)
- Giza: The Tesla Connection: Acoustical Science and the Harvesting of Clean EnergyVon EverandGiza: The Tesla Connection: Acoustical Science and the Harvesting of Clean EnergyNoch keine Bewertungen
- Dark Matter and the Dinosaurs: The Astounding Interconnectedness of the UniverseVon EverandDark Matter and the Dinosaurs: The Astounding Interconnectedness of the UniverseBewertung: 3.5 von 5 Sternen3.5/5 (69)
- Knocking on Heaven's Door: How Physics and Scientific Thinking Illuminate the Universe and the Modern WorldVon EverandKnocking on Heaven's Door: How Physics and Scientific Thinking Illuminate the Universe and the Modern WorldBewertung: 3.5 von 5 Sternen3.5/5 (64)
- A Beginner's Guide to Constructing the Universe: The Mathematical Archetypes of Nature, Art, and ScienceVon EverandA Beginner's Guide to Constructing the Universe: The Mathematical Archetypes of Nature, Art, and ScienceBewertung: 4 von 5 Sternen4/5 (51)
- Lost in Math: How Beauty Leads Physics AstrayVon EverandLost in Math: How Beauty Leads Physics AstrayBewertung: 4.5 von 5 Sternen4.5/5 (125)
- Quantum Spirituality: Science, Gnostic Mysticism, and Connecting with Source ConsciousnessVon EverandQuantum Spirituality: Science, Gnostic Mysticism, and Connecting with Source ConsciousnessBewertung: 4 von 5 Sternen4/5 (6)
- The End of Everything: (Astrophysically Speaking)Von EverandThe End of Everything: (Astrophysically Speaking)Bewertung: 4.5 von 5 Sternen4.5/5 (157)
- Midnight in Chernobyl: The Story of the World's Greatest Nuclear DisasterVon EverandMidnight in Chernobyl: The Story of the World's Greatest Nuclear DisasterBewertung: 4.5 von 5 Sternen4.5/5 (410)
- Bedeviled: A Shadow History of Demons in ScienceVon EverandBedeviled: A Shadow History of Demons in ScienceBewertung: 5 von 5 Sternen5/5 (5)
- Vibration and Frequency: How to Get What You Want in LifeVon EverandVibration and Frequency: How to Get What You Want in LifeBewertung: 4.5 von 5 Sternen4.5/5 (13)
- The Power of Eight: Harnessing the Miraculous Energies of a Small Group to Heal Others, Your Life, and the WorldVon EverandThe Power of Eight: Harnessing the Miraculous Energies of a Small Group to Heal Others, Your Life, and the WorldBewertung: 4.5 von 5 Sternen4.5/5 (54)
- The 60 Minute Quantum Physics Book: Science Made Easy For Beginners Without Math And In Plain Simple EnglishVon EverandThe 60 Minute Quantum Physics Book: Science Made Easy For Beginners Without Math And In Plain Simple EnglishBewertung: 4.5 von 5 Sternen4.5/5 (4)
- Quantum Physics: What Everyone Needs to KnowVon EverandQuantum Physics: What Everyone Needs to KnowBewertung: 4.5 von 5 Sternen4.5/5 (49)
- The Beginning of Infinity: Explanations That Transform the WorldVon EverandThe Beginning of Infinity: Explanations That Transform the WorldBewertung: 5 von 5 Sternen5/5 (60)
- The Magick of Physics: Uncovering the Fantastical Phenomena in Everyday LifeVon EverandThe Magick of Physics: Uncovering the Fantastical Phenomena in Everyday LifeNoch keine Bewertungen
- The Holographic Universe: The Revolutionary Theory of RealityVon EverandThe Holographic Universe: The Revolutionary Theory of RealityBewertung: 4.5 von 5 Sternen4.5/5 (78)
- Let There Be Light: Physics, Philosophy & the Dimensional Structure of ConsciousnessVon EverandLet There Be Light: Physics, Philosophy & the Dimensional Structure of ConsciousnessBewertung: 4.5 von 5 Sternen4.5/5 (57)
- The Illustrated Theory of Everything: The Origin and Fate of the UniverseVon EverandThe Illustrated Theory of Everything: The Origin and Fate of the UniverseBewertung: 5 von 5 Sternen5/5 (1)
- Packing for Mars: The Curious Science of Life in the VoidVon EverandPacking for Mars: The Curious Science of Life in the VoidBewertung: 4 von 5 Sternen4/5 (1396)
- AP Physics 1 Premium, 2024: 4 Practice Tests + Comprehensive Review + Online PracticeVon EverandAP Physics 1 Premium, 2024: 4 Practice Tests + Comprehensive Review + Online PracticeNoch keine Bewertungen