Das könnte Ihnen auch gefallen
- 6th Central Pay Commission Salary CalculatorDokument15 Seiten6th Central Pay Commission Salary Calculatorrakhonde100% (436)
- Crane Gantry Girder (BS5950 - Part1 - 2000)Dokument6 SeitenCrane Gantry Girder (BS5950 - Part1 - 2000)sayed100% (1)
- HX Excel WorkbookDokument14 SeitenHX Excel WorkbookEng/ Abdullah AhmedNoch keine Bewertungen
- Connection DesignDokument33 SeitenConnection Designjesus curielNoch keine Bewertungen
- Coii Niii Cuii and Criii Complexes of Heterocyclic Schiff Base Ligand Synthesis Spectroscopic and Thermal StudyDokument5 SeitenCoii Niii Cuii and Criii Complexes of Heterocyclic Schiff Base Ligand Synthesis Spectroscopic and Thermal StudyIJARP Publications100% (1)
- Shmidt Hammer Method StatementDokument7 SeitenShmidt Hammer Method StatementMurugesan ElaiyappanNoch keine Bewertungen
- API 570 API 571 QuestionsDokument4 SeitenAPI 570 API 571 QuestionsThomas Tucker100% (1)
- Julian Schwinger Selected Papers On Quantum Electrodynamics 1958 PDFDokument443 SeitenJulian Schwinger Selected Papers On Quantum Electrodynamics 1958 PDFLav100% (6)
- Mechanical Test Laboratory Price ListDokument6 SeitenMechanical Test Laboratory Price ListFrancisNoch keine Bewertungen
- Artículos Dos Especies NamaDokument3 SeitenArtículos Dos Especies NamakmiloNoch keine Bewertungen
- Further Alkaloids From Brunsvigia JosephDokument5 SeitenFurther Alkaloids From Brunsvigia JosephPipitNoch keine Bewertungen
- Chalcona e Diidrochalcona PDFDokument3 SeitenChalcona e Diidrochalcona PDFamensetNoch keine Bewertungen
- CH 9790071Dokument18 SeitenCH 9790071Nguyễn Thành VinhNoch keine Bewertungen
- Flavonoides de Lopira AlataDokument3 SeitenFlavonoides de Lopira AlatakmiloNoch keine Bewertungen
- Secotridoides Et Xanthones de Gentiana Burseri 1976 PhytochemistryDokument2 SeitenSecotridoides Et Xanthones de Gentiana Burseri 1976 Phytochemistryasa chiNoch keine Bewertungen
- Two Carotenes and A Prenylated Benzoic Acid Derivative FROM CUDokument6 SeitenTwo Carotenes and A Prenylated Benzoic Acid Derivative FROM CUMiaNoch keine Bewertungen
- 1 s2.0 S0040402013009824 MainDokument5 Seiten1 s2.0 S0040402013009824 MainLizhy PedrazaNoch keine Bewertungen
- Nozaki 2007Dokument3 SeitenNozaki 2007AnaRafaelaSilvaNoch keine Bewertungen
- Triterpene Dienols and Other Constituents From The Bark of Phyllanthus FlexuosusDokument5 SeitenTriterpene Dienols and Other Constituents From The Bark of Phyllanthus FlexuosusTuấn Nguyen AnhNoch keine Bewertungen
- Krukovine, A New Bisbenzylisoquinoline Alkaloid From Abuta SplendidaDokument3 SeitenKrukovine, A New Bisbenzylisoquinoline Alkaloid From Abuta SplendidaDavid ScoNoch keine Bewertungen
- Synthesis of A New Tautomer of Diosphenol BuccocamphorDokument2 SeitenSynthesis of A New Tautomer of Diosphenol BuccocamphorBaban BaidyaNoch keine Bewertungen
- A Diels-Alder-Type Adduct From Artocarpus HeterophyllusDokument3 SeitenA Diels-Alder-Type Adduct From Artocarpus HeterophyllusiswanNoch keine Bewertungen
- Aromatic Compounds From Delphinium Venulosum: in Reoisedform 29 May 1991)Dokument2 SeitenAromatic Compounds From Delphinium Venulosum: in Reoisedform 29 May 1991)Dr-Muhammad Imran TousifNoch keine Bewertungen
- A New Tropane Alkaloid and Other Constituents of Erytorxylum RimosumDokument4 SeitenA New Tropane Alkaloid and Other Constituents of Erytorxylum RimosumGustavo RuizNoch keine Bewertungen
- CoumarinsDokument5 SeitenCoumarinsAmr El DemerdashNoch keine Bewertungen
- Phyto 76 15 991Dokument3 SeitenPhyto 76 15 991JuanManuelAmaroLuisNoch keine Bewertungen
- 102 April2020Dokument7 Seiten102 April2020WINDA TRIA SAFITRINoch keine Bewertungen
- Ab 1032Dokument4 SeitenAb 1032api-19973331Noch keine Bewertungen
- Biflavonoid, Terpen - 1984 - Phytochemistry 23 (2), 323-328Dokument6 SeitenBiflavonoid, Terpen - 1984 - Phytochemistry 23 (2), 323-328Khiem Thai Ba BaoNoch keine Bewertungen
- PW,,H,, DL+ LG.: Scorazanone, A Laza-Anthraqutnone From GonzothalamusDokument3 SeitenPW,,H,, DL+ LG.: Scorazanone, A Laza-Anthraqutnone From GonzothalamusAlexsandro ClaudinoNoch keine Bewertungen
- Phytochemical Analysis, Isolation and Identification of Flavan-3ol From Syrian Pinus HalepensisDokument7 SeitenPhytochemical Analysis, Isolation and Identification of Flavan-3ol From Syrian Pinus HalepensisリファイNoch keine Bewertungen
- Melovinone, An Open Chain Analogue of Melochinone FromDokument2 SeitenMelovinone, An Open Chain Analogue of Melochinone FromwindahNoch keine Bewertungen
- 2001 Isolation of Symlandine From The Roots of Common Comfrey (Symphytum Usin Coutercurrent ChromDokument3 Seiten2001 Isolation of Symlandine From The Roots of Common Comfrey (Symphytum Usin Coutercurrent ChromBackup MelacromNoch keine Bewertungen
- 24-Methylene Tetracyclic Triterpenes From Polyalthia LancilimbaDokument4 Seiten24-Methylene Tetracyclic Triterpenes From Polyalthia LancilimbaamensetNoch keine Bewertungen
- Ijcb 45B (11) 2574-2579Dokument6 SeitenIjcb 45B (11) 2574-2579Deepshikha GuptaNoch keine Bewertungen
- Baytop, T (1984) In: A Flavonol Glycoside From Lysimachla A4AurltianaDokument3 SeitenBaytop, T (1984) In: A Flavonol Glycoside From Lysimachla A4AurltianaMeylianaNoch keine Bewertungen
- Triterpenoids and Chalcone From Syzygium SamarangenseDokument3 SeitenTriterpenoids and Chalcone From Syzygium SamarangenseMinyty LeNoch keine Bewertungen
- Alam G DKK 2005 - Structure Elucidation of Bioactive Alkaloid Compounds Isolated From Sponge Petrosia SPDokument5 SeitenAlam G DKK 2005 - Structure Elucidation of Bioactive Alkaloid Compounds Isolated From Sponge Petrosia SPSeptynelya ThenuNoch keine Bewertungen
- Likhitwitayawuid 1993Dokument11 SeitenLikhitwitayawuid 1993drirene2022Noch keine Bewertungen
- The Structure and Stereochemistry of Artemin": (Revised Received 14 March 1977)Dokument2 SeitenThe Structure and Stereochemistry of Artemin": (Revised Received 14 March 1977)JuanManuelAmaroLuisNoch keine Bewertungen
- Pregnane Glycosides From Sansevieria Trifasciata: OH J,, K J - . JDokument5 SeitenPregnane Glycosides From Sansevieria Trifasciata: OH J,, K J - . JRommelAnastacioNoch keine Bewertungen
- Deve HatDokument5 SeitenDeve HatOkky Winang SaktyawanNoch keine Bewertungen
- A Novel 2-Hydroxyflavanone From Collinsonia CanadensisDokument3 SeitenA Novel 2-Hydroxyflavanone From Collinsonia CanadensisAuroTest. deNoch keine Bewertungen
- 1 s2.0 S003194220200184X MainDokument4 Seiten1 s2.0 S003194220200184X MainanneNoch keine Bewertungen
- Journal of Electroanalytical Chemistry: Habibe Tezcan, Elif Uzluk, Mehmet Levent AksuDokument12 SeitenJournal of Electroanalytical Chemistry: Habibe Tezcan, Elif Uzluk, Mehmet Levent AksuRobert WinterNoch keine Bewertungen
- C27H4fi03: T., F., E., H., R. S., 17Dokument17 SeitenC27H4fi03: T., F., E., H., R. S., 17Phil DinningNoch keine Bewertungen
- Isolation and Identification of Alkaloids From Croton LobatusDokument4 SeitenIsolation and Identification of Alkaloids From Croton LobatusUtari Putri ErinaNoch keine Bewertungen
- Briononic Acid From The Hexane Extract of Sandoricum Koetjape MERR STEM BARK (Meliaceae)Dokument3 SeitenBriononic Acid From The Hexane Extract of Sandoricum Koetjape MERR STEM BARK (Meliaceae)Winda ApriyentiNoch keine Bewertungen
- Dinuclear Rhodium (II1) Complexes: Synthesis and Crystal and Molecular Structures Two Amine-Oxime Complexes of Rhodium (Dokument5 SeitenDinuclear Rhodium (II1) Complexes: Synthesis and Crystal and Molecular Structures Two Amine-Oxime Complexes of Rhodium (Arijit dasguptaNoch keine Bewertungen
- A 7-Hydroxyaporphine Alkaloid From Desmos DasymachalusDokument2 SeitenA 7-Hydroxyaporphine Alkaloid From Desmos DasymachalusamensetNoch keine Bewertungen
- Jurnal 8 Struktur Baru Dalam Piper Retrofractum 2013Dokument5 SeitenJurnal 8 Struktur Baru Dalam Piper Retrofractum 2013Muharrom RiezkyNoch keine Bewertungen
- Biflavonoids From Lonicera Japonica - PhytochemistryDokument5 SeitenBiflavonoids From Lonicera Japonica - PhytochemistryTàiNguyễnThànhNoch keine Bewertungen
- Taraxastane Glycosides From Eclipta AlbaDokument5 SeitenTaraxastane Glycosides From Eclipta AlbaMinyty LeNoch keine Bewertungen
- Peracid Oxidation of Amines To NitroalkanesDokument3 SeitenPeracid Oxidation of Amines To NitroalkanesSunny ChosaNoch keine Bewertungen
- Molecules 12 01796Dokument9 SeitenMolecules 12 01796Kalpesh PatelNoch keine Bewertungen
- Isolation of (-) - Epicatechin From Trichilia Emetica Whole SeedsDokument5 SeitenIsolation of (-) - Epicatechin From Trichilia Emetica Whole Seedstuấn anhNoch keine Bewertungen
- Hetero Cyclic Compounds Mixed Book NotesDokument25 SeitenHetero Cyclic Compounds Mixed Book NotesSundas FatimaNoch keine Bewertungen
- Phytotoxic and Nematicidal Components of Lavandula LuisieriDokument6 SeitenPhytotoxic and Nematicidal Components of Lavandula Luisieriluis fernando julio torresNoch keine Bewertungen
- Notes: A New Alkaloid From Two Coccinellid Beetles Harmonia Axyridis and Aiolocaria HexaspilotaDokument3 SeitenNotes: A New Alkaloid From Two Coccinellid Beetles Harmonia Axyridis and Aiolocaria HexaspilotaSupriono ChinagaNoch keine Bewertungen
- Articulo 4Dokument5 SeitenArticulo 4Viviana TorresNoch keine Bewertungen
- Sitosterol Stigmasterol 2-Hydroxy-Hexadecanoic AcidDokument7 SeitenSitosterol Stigmasterol 2-Hydroxy-Hexadecanoic AcidtoanphandlNoch keine Bewertungen
- Atkinson05 1Dokument15 SeitenAtkinson05 1Shing Cho MaNoch keine Bewertungen
- No Card IcinDokument12 SeitenNo Card IcinHazrati UmmiNoch keine Bewertungen
- 6-Methoxybenzoxazolinone and Triterpenoids From Roots of Scoparia DulcisDokument3 Seiten6-Methoxybenzoxazolinone and Triterpenoids From Roots of Scoparia DulcisMinyty LeNoch keine Bewertungen
- Balashova TV - Lanthanide Complexes With The Schiff Base Containing Sterically Hindered Phenol Synthesis Structure and Luminescence Properties - 2017Dokument6 SeitenBalashova TV - Lanthanide Complexes With The Schiff Base Containing Sterically Hindered Phenol Synthesis Structure and Luminescence Properties - 2017Iuliana FloreaNoch keine Bewertungen
- Chirality Organization of Aniline Oligomers Through Hydrogen Bonds of Amino Acid MoietiesDokument4 SeitenChirality Organization of Aniline Oligomers Through Hydrogen Bonds of Amino Acid MoietiesDiogomussumNoch keine Bewertungen
- 25th International Congress of Pure and Applied Chemistry: Plenary Lectures Presented at the 25th International Congress of Pure and Applied Chemistry, Jerusalem, Israel 6–11 July 1975Von Everand25th International Congress of Pure and Applied Chemistry: Plenary Lectures Presented at the 25th International Congress of Pure and Applied Chemistry, Jerusalem, Israel 6–11 July 1975Bewertung: 5 von 5 Sternen5/5 (1)
- Carbohydrate Chemistry—8: Plenary Lectures Presented at the Eighth International Symposium on Carbohydrate Chemistry, Kyoto, Japan 16 - 20 August 1976Von EverandCarbohydrate Chemistry—8: Plenary Lectures Presented at the Eighth International Symposium on Carbohydrate Chemistry, Kyoto, Japan 16 - 20 August 1976K. OnoderaNoch keine Bewertungen
- European Journal of PhycologyDokument15 SeitenEuropean Journal of PhycologyamensetNoch keine Bewertungen
- European Journal of PhycologyDokument11 SeitenEuropean Journal of PhycologyamensetNoch keine Bewertungen
- A Chlorophyll Fluorescence Index To Estimate Short-Term Rates of Photosynthesis by Intertidal MicrophytobenthosDokument14 SeitenA Chlorophyll Fluorescence Index To Estimate Short-Term Rates of Photosynthesis by Intertidal MicrophytobenthosamensetNoch keine Bewertungen
- Photosynthetic Activity of Intertidal Microphytobenthic Communities During Emersion: in Situ Measurements of Chlorophyll Fluorescence (Pam) and Co Flux (Irga)Dokument10 SeitenPhotosynthetic Activity of Intertidal Microphytobenthic Communities During Emersion: in Situ Measurements of Chlorophyll Fluorescence (Pam) and Co Flux (Irga)amensetNoch keine Bewertungen
- Art:10 1007/BF02183042Dokument7 SeitenArt:10 1007/BF02183042amensetNoch keine Bewertungen
- (2004) - (Suggett Et Al.) - Evaluation of Biophysical and Optical Determination of Light Absortion by Photpsystem II in PhytoplanktonDokument17 Seiten(2004) - (Suggett Et Al.) - Evaluation of Biophysical and Optical Determination of Light Absortion by Photpsystem II in PhytoplanktonamensetNoch keine Bewertungen
- Influence of Nitrate Feeding On Carbon Dioxide Fixation by MicroalgaeDokument13 SeitenInfluence of Nitrate Feeding On Carbon Dioxide Fixation by MicroalgaeamensetNoch keine Bewertungen
- (2008) - (Rech Et Al.) - Carbon Fixation and Carbonic Anhydrase ActivityDokument7 Seiten(2008) - (Rech Et Al.) - Carbon Fixation and Carbonic Anhydrase ActivityamensetNoch keine Bewertungen
- (1997) - (De Las Rivas) - Structure and Thermal Stability of Photosystem II ReactionDokument7 Seiten(1997) - (De Las Rivas) - Structure and Thermal Stability of Photosystem II ReactionamensetNoch keine Bewertungen
- Nitella Cernua (Characeae, Chlorophyta) : Photosynthetic Characteristics of A Tropical Population ofDokument10 SeitenNitella Cernua (Characeae, Chlorophyta) : Photosynthetic Characteristics of A Tropical Population ofamensetNoch keine Bewertungen
- (2008) - (Rech Et Al.) - Carbon Fixation and Carbonic Anhydrase ActivityDokument7 Seiten(2008) - (Rech Et Al.) - Carbon Fixation and Carbonic Anhydrase ActivityamensetNoch keine Bewertungen
- (2005) - (Ralph and Gademann) - Rapid Light Curves - A Powerful Tool To Asses Photosynthetic ActivityDokument16 Seiten(2005) - (Ralph and Gademann) - Rapid Light Curves - A Powerful Tool To Asses Photosynthetic ActivityamensetNoch keine Bewertungen
- (2006) - (Ban Et Al.)Dokument18 Seiten(2006) - (Ban Et Al.)amensetNoch keine Bewertungen
- (2004) - (Necchi) - Light-Related Photosynthetic Characteristics of Lotic MacroalgaeDokument17 Seiten(2004) - (Necchi) - Light-Related Photosynthetic Characteristics of Lotic MacroalgaeamensetNoch keine Bewertungen
- (2004) - (Necchi) - Light-Related Photosynthetic Characteristics of Lotic MacroalgaeDokument17 Seiten(2004) - (Necchi) - Light-Related Photosynthetic Characteristics of Lotic MacroalgaeamensetNoch keine Bewertungen
- (2004) - (Suggett Et Al.) - Evaluation of Biophysical and Optical Determination of Light Absortion by Photpsystem II in PhytoplanktonDokument17 Seiten(2004) - (Suggett Et Al.) - Evaluation of Biophysical and Optical Determination of Light Absortion by Photpsystem II in PhytoplanktonamensetNoch keine Bewertungen
- (2005) - (Ralph and Gademann) - Rapid Light Curves - A Powerful Tool To Asses Photosynthetic ActivityDokument16 Seiten(2005) - (Ralph and Gademann) - Rapid Light Curves - A Powerful Tool To Asses Photosynthetic ActivityamensetNoch keine Bewertungen
- (1997) - (Sakshaug, 1997) - Parameters of PhotosynthesisDokument34 Seiten(1997) - (Sakshaug, 1997) - Parameters of PhotosynthesisamensetNoch keine Bewertungen
- (2004) - (Necchi) - Light-Related Photosynthetic Characteristics of Lotic MacroalgaeDokument17 Seiten(2004) - (Necchi) - Light-Related Photosynthetic Characteristics of Lotic MacroalgaeamensetNoch keine Bewertungen
- (1996) - (Herrmann, 1996) - Effects of UV Radiation On Photosynthesis of PhytoplanktonDokument8 Seiten(1996) - (Herrmann, 1996) - Effects of UV Radiation On Photosynthesis of PhytoplanktonamensetNoch keine Bewertungen
- (1997) - (Yamane Et Al.) - Increases in The Fluorescence Fo Level and Reversible InhibitionDokument8 Seiten(1997) - (Yamane Et Al.) - Increases in The Fluorescence Fo Level and Reversible InhibitionamensetNoch keine Bewertungen
- (1998) - (Beer Et Al.) - Measuring Photosynthetic Rates in Seagrasses by Pulse Amplitude Modulated (PAM) FluorometryDokument8 Seiten(1998) - (Beer Et Al.) - Measuring Photosynthetic Rates in Seagrasses by Pulse Amplitude Modulated (PAM) FluorometryamensetNoch keine Bewertungen
- (1997) - (Oxborough and Baker) - Resolving Chlorophyll A Fluorescence Images of Photosynthetic Efficiency Into Photochemical and Non-Photochemical Components 1Dokument8 Seiten(1997) - (Oxborough and Baker) - Resolving Chlorophyll A Fluorescence Images of Photosynthetic Efficiency Into Photochemical and Non-Photochemical Components 1amensetNoch keine Bewertungen
- The Effects of Anabaena Spiroides (Cyanophyceae) Exopolysaccharide On Copper Toxicity To Simocephalus Serrulatus (Cladocera, Daphnidae)Dokument8 SeitenThe Effects of Anabaena Spiroides (Cyanophyceae) Exopolysaccharide On Copper Toxicity To Simocephalus Serrulatus (Cladocera, Daphnidae)amensetNoch keine Bewertungen
- (1995) - (Barón, Arellano and Gorgé) - Copper and Photosystem IIDokument25 Seiten(1995) - (Barón, Arellano and Gorgé) - Copper and Photosystem IIamensetNoch keine Bewertungen
- (1991) - (Havaux, Strasser and Greppin) - A Theoretical and Experimental Analysis of The QT, and QN CoefficientsDokument15 Seiten(1991) - (Havaux, Strasser and Greppin) - A Theoretical and Experimental Analysis of The QT, and QN CoefficientsamensetNoch keine Bewertungen
- (1990) - (Kooten and Snel) - The Use of Chlorophyll Fluorescence NomenclatureDokument4 Seiten(1990) - (Kooten and Snel) - The Use of Chlorophyll Fluorescence NomenclatureamensetNoch keine Bewertungen
- Walseng - Simocephalus SerrulatusDokument1 SeiteWalseng - Simocephalus SerrulatusamensetNoch keine Bewertungen
- Astaxanthinnonconf 2Dokument24 SeitenAstaxanthinnonconf 2amensetNoch keine Bewertungen
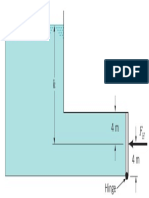
- Fluid Statics: F I G U R E P2.69Dokument1 SeiteFluid Statics: F I G U R E P2.69andres bernalNoch keine Bewertungen
- Oxo-Degradable Additive: GeneralDokument4 SeitenOxo-Degradable Additive: GeneralLaili AzkiyahNoch keine Bewertungen
- Qpedia Apr09 Basic Principles of Wind Tunnel Design9Dokument3 SeitenQpedia Apr09 Basic Principles of Wind Tunnel Design9Ryan FadhliNoch keine Bewertungen
- Unclassified Ad Number Limitation Changes TODokument36 SeitenUnclassified Ad Number Limitation Changes TORuben's OscarNoch keine Bewertungen
- Lesson 6.2 Chemical ReactionsDokument20 SeitenLesson 6.2 Chemical Reactions16394-Karm M. Basel KabbaniNoch keine Bewertungen
- Primal Et Al. 2004Dokument13 SeitenPrimal Et Al. 2004Francisco OppsNoch keine Bewertungen
- Writing A UMAT - 0Dokument18 SeitenWriting A UMAT - 0Noushad Bin Jamal Structural EngineerNoch keine Bewertungen
- Combined Set12Dokument159 SeitenCombined Set12Nguyễn Sơn LâmNoch keine Bewertungen
- FRC1Dokument49 SeitenFRC1Pavan Kalyan YadavNoch keine Bewertungen
- Heat Exchanger Question - ANSDokument7 SeitenHeat Exchanger Question - ANSBikas SahaNoch keine Bewertungen
- 3) M1 Dynamics of A Particle Moving in A Straight LineDokument84 Seiten3) M1 Dynamics of A Particle Moving in A Straight LineisamalhassanNoch keine Bewertungen
- g7 3j04 Lab - FinalDokument46 Seiteng7 3j04 Lab - Finalyliu8877100% (1)
- Lesson 1 - Solid, Liquid, and Gas (Grade 3)Dokument26 SeitenLesson 1 - Solid, Liquid, and Gas (Grade 3)Christian TabalanNoch keine Bewertungen
- Finite Element Analysis of Soil Bearing Capacity Using PlaxisDokument5 SeitenFinite Element Analysis of Soil Bearing Capacity Using PlaxisRehan HakroNoch keine Bewertungen
- Physics PracticalsDokument108 SeitenPhysics PracticalsASHISH KHUTENoch keine Bewertungen
- Energy Efficiency in Pump SpecificationDokument8 SeitenEnergy Efficiency in Pump SpecificationFuad Al-AwzariNoch keine Bewertungen
- Line Data: Piping Isometric DrawingsDokument1 SeiteLine Data: Piping Isometric Drawingsksat85Noch keine Bewertungen
- Analysis and Design of Multi-Storeyed Building by Steel Concrete Composite StructureDokument9 SeitenAnalysis and Design of Multi-Storeyed Building by Steel Concrete Composite StructureUsha EngineeringNoch keine Bewertungen
- Header BowmanDokument12 SeitenHeader BowmanMehrdad SakhaieNoch keine Bewertungen
- F - UNIT 2 - Engg Mecha Statics - Class 13Dokument19 SeitenF - UNIT 2 - Engg Mecha Statics - Class 13Govardhan RaoNoch keine Bewertungen
- Physics I Projectile Motion Practice Problems 4 1 SolnDokument16 SeitenPhysics I Projectile Motion Practice Problems 4 1 SolnFranceNoch keine Bewertungen
- Climatic Factor For Design of BuildingDokument66 SeitenClimatic Factor For Design of Buildingarchitectneha614Noch keine Bewertungen
- Derivation of The Schrodinger Equation From Newtonian MechanicsDokument7 SeitenDerivation of The Schrodinger Equation From Newtonian MechanicsCyrille TijsselingNoch keine Bewertungen